Defect Thermodynamics & Doping
Tip
You can run this notebook interactively through Google Colab or Binder – just click the links! (Then uncomment the !pip install... cell).
# # If running on Colab; uncomment to install doped, clone the repo to download the example data and cd in to examples folder:
# !pip install doped
# !git clone https://github.com/SMTG-Bham/doped
# %cd doped/examples
%matplotlib inline
from monty.serialization import loadfn
CdTe_thermo = loadfn("CdTe/CdTe_LZ_thermo_wout_meta.json.gz") # load our DefectThermodynamics object
Let’s first check the calculated symmetries and degeneracy factors of our defects:
CdTe_thermo.get_symmetries_and_degeneracies()
| Site_Symm | Defect_Symm | g_Orient | g_Spin | g_Total | Mult | ||
|---|---|---|---|---|---|---|---|
| Defect | q | ||||||
| v_Cd | 0 | Td | C2v | 6.0 | 1.0 | 6.0 | 1.0 |
| -1 | Td | C3v | 4.0 | 2.0 | 8.0 | 1.0 | |
| -2 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 | |
| v_Te | +2 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 |
| +1 | Td | Td | 1.0 | 2.0 | 2.0 | 1.0 | |
| 0 | Td | C2v | 6.0 | 1.0 | 6.0 | 1.0 | |
| -1 | Td | Cs | 12.0 | 2.0 | 24.0 | 1.0 | |
| -2 | Td | Cs | 12.0 | 1.0 | 12.0 | 1.0 | |
| Cd_Te | +2 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 |
| +1 | Td | C2v | 6.0 | 2.0 | 12.0 | 1.0 | |
| 0 | Td | Cs | 12.0 | 1.0 | 12.0 | 1.0 | |
| -1 | Td | Td | 1.0 | 4.0 | 4.0 | 1.0 | |
| -2 | Td | C1 | 24.0 | 1.0 | 24.0 | 1.0 | |
| Te_Cd | +2 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 |
| +1 | Td | C3v | 4.0 | 2.0 | 8.0 | 1.0 | |
| 0 | Td | C3v | 4.0 | 1.0 | 4.0 | 1.0 | |
| -1 | Td | Cs | 12.0 | 2.0 | 24.0 | 1.0 | |
| -2 | Td | Cs | 12.0 | 1.0 | 12.0 | 1.0 | |
| Cd_i_Td_Cd2.83 | +2 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 |
| +1 | Td | Td | 1.0 | 2.0 | 2.0 | 1.0 | |
| 0 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 | |
| Cd_i_Td_Te2.83 | +2 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 |
| +1 | Td | Td | 1.0 | 2.0 | 2.0 | 1.0 | |
| 0 | Td | Td | 1.0 | 1.0 | 1.0 | 1.0 | |
| Te_i_Td_Te2.83 | +2 | C1 | Cs | 0.5 | 1.0 | 0.5 | 24.0 |
| +1 | Cs | C2v | 0.5 | 2.0 | 1.0 | 12.0 | |
| 0 | C1 | C2 | 0.5 | 1.0 | 0.5 | 24.0 | |
| -1 | C1 | C2 | 0.5 | 2.0 | 1.0 | 24.0 | |
| -2 | C1 | C2 | 0.5 | 1.0 | 0.5 | 24.0 |
Tip
As noted in the DefectThermodynamics.get_symmetries_and_degeneracies() docstring, the multiplicity (Mult) output here is given with respect to the primitive unit cell.
For interstitials, the bulk site symmetry corresponds to the point symmetry of the interstitial site with no relaxation of the host structure, while for vacancies/substitutions it is simply the symmetry of their corresponding bulk site. The defect symmetry (Defect_Symm) corresponds to the final relaxed defect symmetry.
The product of the defect degeneracy factors (such as orientational and spin, which are automatically computed by doped) and site multiplicity determine the pre-factor is used in the calculation of defect/carrier concentrations (and thus doping / Fermi level behaviour). For more explanation and discussion of this, see e.g. Impact of metastable defect structures on carrier recombination in solar cells, [Guidelines for robust and reproducible point defect simulations in crystals]https://doi.org/10.1038/s41578-025-00879-y), Imperfections are not 0 K: free energy of point defects in crystals.
Note
Note: doped tries to use the defect_entry.defect_supercell to determine the relaxed site symmetry. However, it should be noted that this is not guaranteed to work in all cases; namely for certain non-trivial supercell expansion matrices (e.g. a 2x1x2 expansion)(particularly with high-symmetry materials) which can mess up the periodicity of the cell. doped tries to automatically check if this is the case, and will warn you if so.
If periodicity-breaking does prevent auto-symmetry determination, you can manually determine the relaxed defect and bulk-site point symmetries, and/or orientational degeneracy, from visualising the structures (e.g. using VESTA)(can use get_orientational_degeneracy to obtain the corresponding orientational degeneracy factor for given defect/bulk-site point symmetries) and setting the corresponding values in the calculation_metadata['relaxed point symmetry']/['bulk site symmetry'] and/or degeneracy_factors['orientational degeneracy'] attributes.
Let’s see what our defect formation energy diagram looks like:
CdTe_thermo.dist_tol = 1.6 # expand distance tolerance (default = 1.6 Å) for grouping defect entries of the same type (Te_i in this case); see https://doped.readthedocs.io/en/latest/plotting_customisation_tutorial.html#dist-tol
plot = CdTe_thermo.plot(limit="Te-rich")
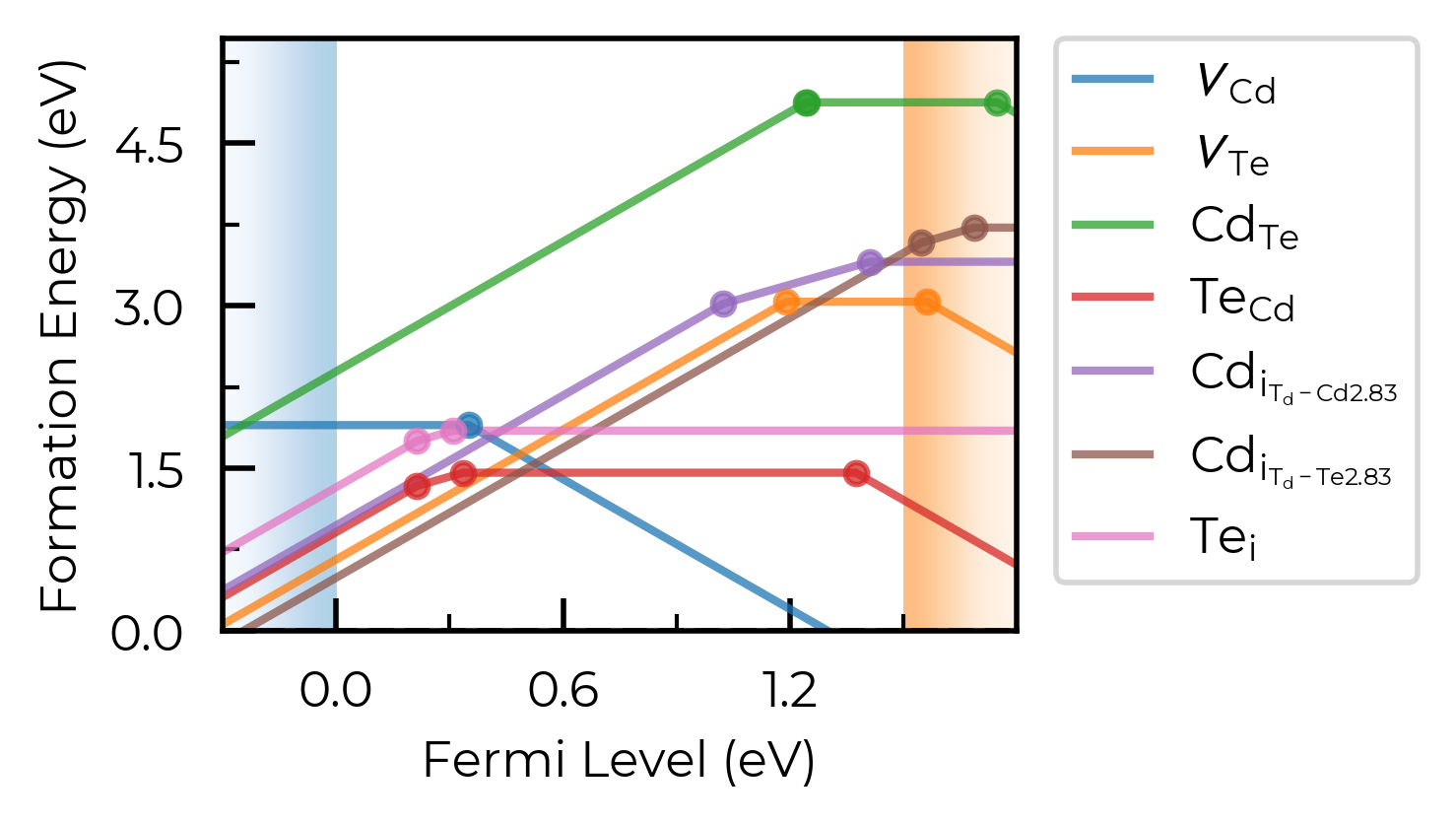
Tip
As shown above, can specify the chemical potential limit at which to obtain and plot the defect formation energies using the limit parameter, which we can set to either "X-rich"/"X-poor" where X is an element in the system, in which case the most X-rich/poor limit will be used (e.g. “Cd-rich”), or a key in the chempots["limits"] dictionary (e.g. "Cd-CdTe" from that shown above). Alternatively, one can also provide a single chemical potential limit in the form of a dictonary to the DefectThermodynamics methods – see docstrings for more details.
Note
Note that in doped, the "band_gap" and "vbm" values in DefectEntry.calculation_metadata are used to determine the position of the band edges in the defect formation energy plots and doping window / dopability limit functions, and the reference value (with the VBM at 0 eV) of any reported Fermi levels. These values are automatically taken from the bulk supercell calculation during defect parsing, but if for any reason the band edges in your bulk supercell calculation are not the correct values (e.g. true VBM/CBM not included in the k-point grid), then the bulk_band_gap_vr parameter should be provided during defect parsing so that "band_gap" and "vbm" are extracted from it (see docstring / API docs for info).
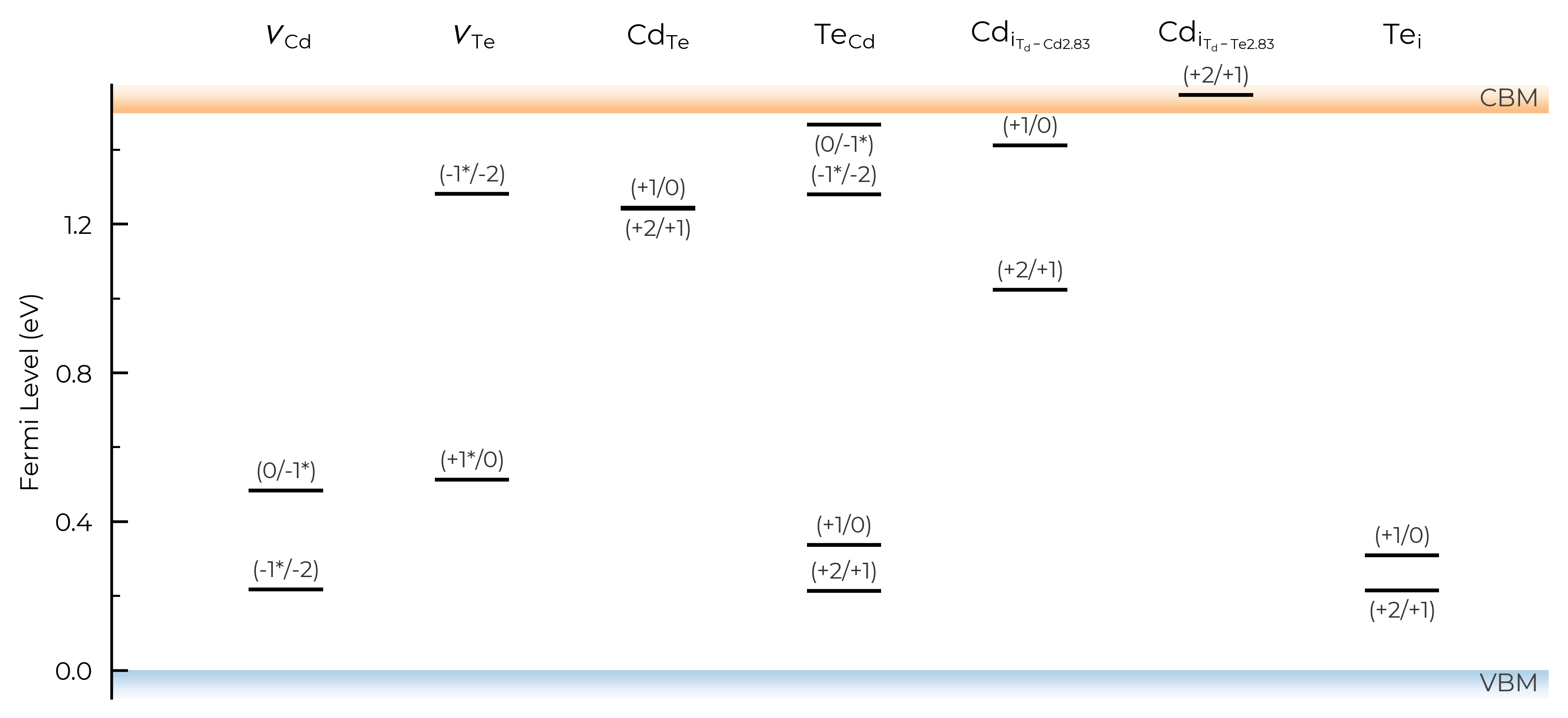
We can also visualise the charge transition levels directly using the plot_transition_levels() method:
tl_fig = CdTe_thermo.plot_transition_levels()
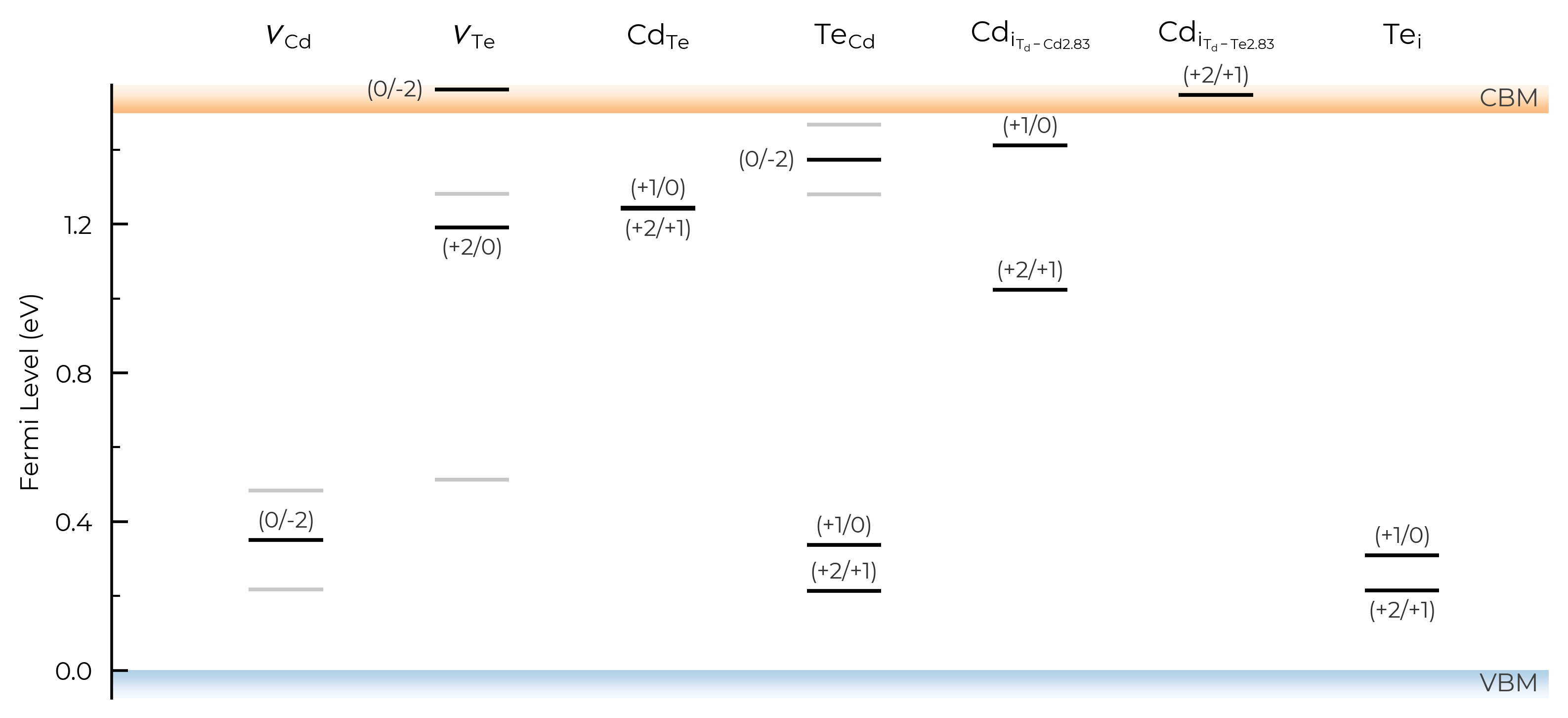
As with the other get_transition_levels() and print_transition_levels() DefectThermodynamics methods, we can set all=True to include single-electron transitions involving metastable charge states, which are denoted with a * in the labels (and which can be important for recombination, migration, defect dynamics and concentrations; see e.g. Faraday Discuss 2022, Phys Rev B 2016, Phys Rev Mater 2024, Phys Rev Mater 2021, or Chem Soc Rev 2023).
tl_fig_all = CdTe_thermo.plot_transition_levels(all=True) # include metastable TLs
# see the plotting customisation tutorial for more TL plotting options!
Dopability Limits
As a first analysis of the dopability of our system, we can use the get_doability_limits() and get_doping_windows() methods of the DefectThermodynamics object, to determine our n/p-type doping limits:
CdTe_thermo.get_dopability_limits()
| Limit | Compensating Defect | Dopability Limit (eV from VBM/CBM) | |
|---|---|---|---|
| p-type | Te-rich (CdTe-Te) | Cd_i_Td_Te2.83_+2 | -0.244 |
| n-type | Cd-rich (Cd-CdTe) | v_Cd_-2 | 1.925 |
CdTe_thermo.get_doping_windows()
| Limit | Compensating Defect | Doping Window (eV at VBM/CBM) | |
|---|---|---|---|
| p-type | Te-rich (CdTe-Te) | Cd_i_Td_Te2.83_+2 | 0.489 |
| n-type | Cd-rich (Cd-CdTe) | v_Cd_-2 | 0.853 |
These values are explained in more detail in the docstrings, but essentially they give us a rough estimate of the p/n-type dopability of our system, based on the native defect thermodynamics. The dopability limits are defined as the Fermi level positions at which the lowest-energy compensating defect becomes zero, while the doping windows are the energies of the lowest-energy compensating defects at the corresponding band-edge (VBM/CBM).
Tip
A large doping window or a dopability limit far from the band-edge position indicates high dopability for that carrier type (i.e. that the system is likely easier to dope p/n-type).
For example here we see that \(Cd_{i}^{+2}\) (coordinated by Te anions) is our dominant (lowest-energy) compensating native donor under p-type (Te-rich) conditions, and we can see the doping window of 0.49 eV corresponds to its formation energy at the VBM under Te-rich conditions in the plot above. Meanwhile, \(V_{Cd}^{-2}\) is our dominant native compensating acceptor under n-type (Cd-rich) conditions. From this initial analysis, we can see that our native defect thermodynamics suggests that CdTe is more n-type dopable than p-type dopable (having alarger n-type doping window/limit).
Doping Calculations
The above analysis can give a useful qualitative picture of the expected dopability of our material, but we can go on to directly calculate the predicted defect/carrier concentrations and Fermi level positions under various conditions.
To quantitatively analyse the electronic (doping) behaviour of defects in our material, we need the electronic density of states (DOS) of the bulk material. Usually this is performed as a static calculation of the primitive cell, with dense k-point sampling for a converged DOS (see vaspup2.0; recommended to use a large NEDOS and ISMEAR = -5) and an accurate band gap (using SOC etc if necessary). The DOS band gap / VBM should match that of your DefectThermodynamics object/plot (doped will throw a warning if it detects that they differ significantly). See the DOS Section on the Tips page for advice on this calculation.
The bulk_dos parameter is used to provide our calculated DOS in the doped thermodynamics functions. We can provide bulk_dos during DefectThermodynamics initialisation, with DefectsParser.get_defect_thermodynamics(), or by setting the bulk_dos property (as shown below). Alternatively, we can provide the bulk_dos input to the DefectThermodynamics Fermi level / concentration methods, but this means re-parsing the DOS object each time which may be slow. With all cases, bulk_dos can be provided as a path to a vasprun.xml(.gz) output (of a DOS calculation), a Vasprun object, or a pymatgen FermiDos object.
# provide the bulk DOS for Fermi level / carrier concentration / defect concentration calculations:
CdTe_thermo.bulk_dos = "CdTe/CdTe_prim_k181818_NKRED_2_vasprun.xml.gz"
Let’s first plot the carrier concentrations versus Fermi level in our material, to see what sort of Fermi level position we would need to obtain a target free carrier concentration etc. Here for CdTe, we’ll plot these quantities at 300K (room temperature) and 875K (a typical annealing temperature):
import matplotlib.pyplot as plt
import numpy as np
f, ax = plt.subplots(figsize=(5, 3))
band_gap = CdTe_thermo.bulk_dos.get_gap(tol=1e-6)
anneal_T = 875
e_concs = []; h_concs = []; fermi_levels = np.linspace(-0.3, band_gap + 0.3, 100)
high_temp_e_concs = []; high_temp_h_concs = []
for fermi_level in fermi_levels:
e_conc, h_conc = CdTe_thermo.bulk_dos.get_e_h_concs(
fermi_level=CdTe_thermo.vbm + fermi_level, temperature=300)
e_concs.append(e_conc); h_concs.append(h_conc)
e_conc, h_conc = CdTe_thermo.bulk_dos.get_e_h_concs(
fermi_level=CdTe_thermo.vbm + fermi_level, temperature=anneal_T)
high_temp_e_concs.append(e_conc); high_temp_h_concs.append(h_conc)
ax.plot(fermi_levels, h_concs, label="$[p]$ ($T$ = 300 K)", color="C0")
ax.plot(fermi_levels, high_temp_h_concs, label=f"$[p]$ ($T$ = {anneal_T} K)", color="C0", ls="--", alpha=0.5)
ax.plot(fermi_levels, e_concs, label="$[n]$ ($T$ = 300 K)", color="C1")
ax.plot(fermi_levels, high_temp_e_concs, label=f"$[n]$ ($T$ = {anneal_T} K)", color="C1", ls="--", alpha=0.5)
ax.set_xlabel("Fermi Level (eV)"); ax.set_ylabel("Concentration (cm$^{-3}$)")
ax.legend(); ax.semilogy()
# show VB in blue from -0.3 to 0 eV and CB in orange from CBM to CBM + 0.3 eV:
ax.imshow([(0, 1), (0, 1)], cmap=plt.cm.Blues, extent=(ax.get_xlim()[0], 0, ax.get_ylim()[1], ax.get_ylim()[0]),
vmin=0, vmax=3, interpolation="bicubic", rasterized=True, aspect="auto")
ax.imshow([(1, 0), (1, 0)], cmap=plt.cm.Oranges, extent=(band_gap, band_gap + 0.3, ax.get_ylim()[0], ax.get_ylim()[1]),
vmin=0, vmax=3, interpolation="bicubic", rasterized=True, aspect="auto")
ax.set_xlim(-0.3, band_gap + 0.3); ax.set_ylim(1e10, 1e21); # set axis limits
f
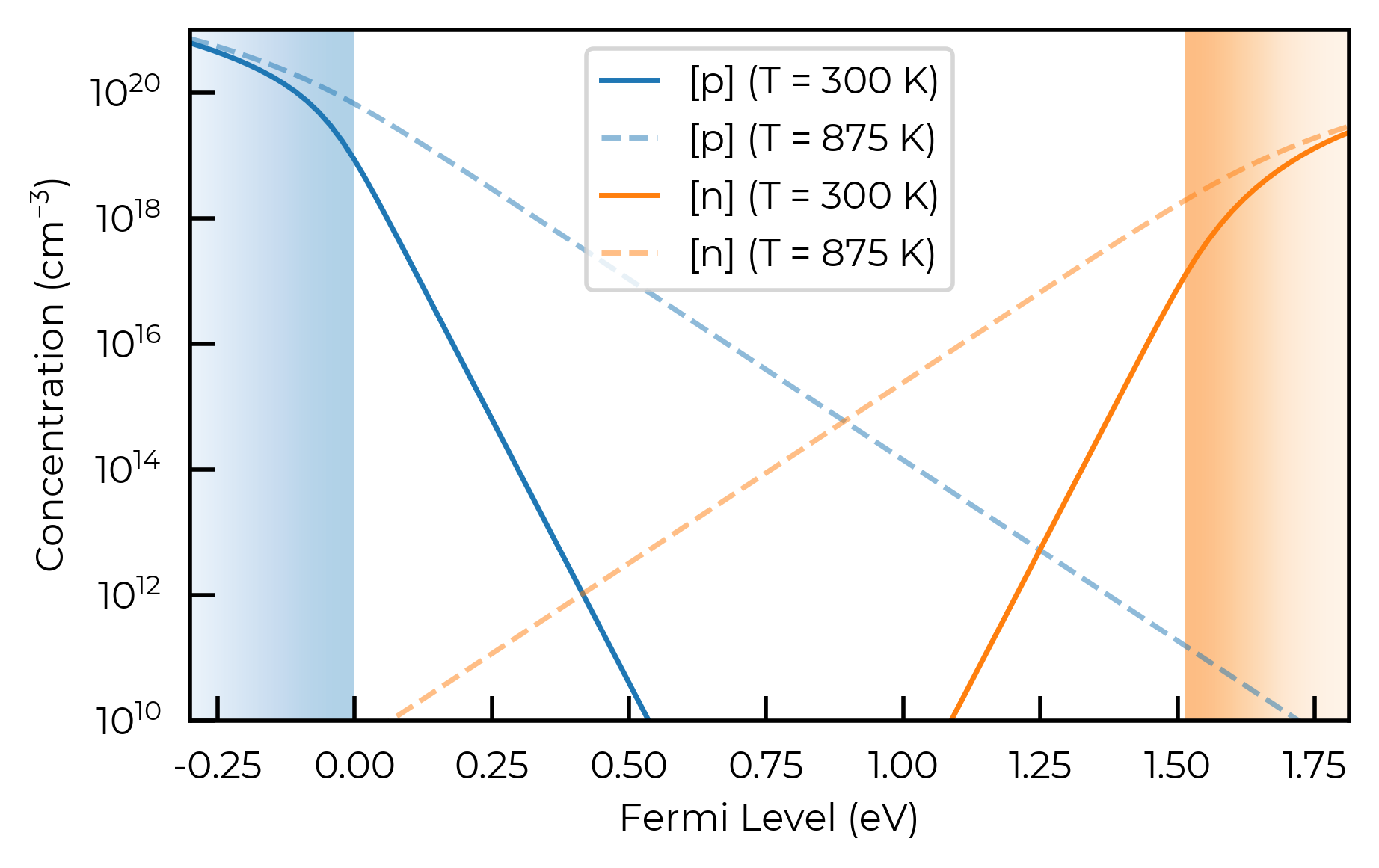
Here we can see a clear disparity in the n-type and p-type carrier concentrations, where the hole concentration is significantly higher than the electron concentration for the same energy separation of the Fermi level with the corresponding band edge (VBM/CBM), which is a direct result of the significantly higher DOS at the VBM in CdTe compared to the CBM.
Tip
By tracking the Fermi level & carrier concentration during annealing and upon cooling, this plot above can be used to analyse the type of compensation occurring for a given system (i.e. electronic, ionic or mixed). Given the annealing Fermi level & carrier concentration as a starting point, upon cooling it will either move horizontally to retain the carrier concentration while shifting the Fermi level closer to the band edge for full electronic compensation, move vertically downwards to decrease carrier concentrations at the same/similar Fermi level if you have self-compensation at that Fermi level, or somewhere in-between for compensation by other defects / self-compensation at a different Fermi level position / mixed electronic-ionic compensation.
CdTe: Cd-Rich
Let’s first look at the carrier and defect calculations in CdTe at the Cd-rich (i.e. most n-type) chemical potential limit, as a function of the device annealing/growth temperature.
The formation energies of charged defects in a material depend on the electronic chemical potential (i.e. the Fermi level), which in turn depends on the net population of all charged defects in the system. The equilibrium concentrations of charged point defects are therefore mutually dependent, and so the equilibrium Fermi level position must be solved self-consistently. Typically this is done by imposing the constraint of net charge-neutrality, and solving for the Fermi level which satisfies this condition – as implemented in doped.
Note
In most cases, materials are synthesised or processed under elevated temperatures (‘annealing’), before being cooled (‘quenched’) to the device operating temperature. Due to the exponential temperature dependence of equilibrium defect concentration and greater availability of thermal energy, it is generally expected that most defects will be created during the initial high-temperature annealing step, with their total concentration then remaining fixed due to kinetic trapping upon cooling — this being known as the ‘frozen defect approximation’.
While the total concentration of each defect is assumed to be fixed upon cooling within this approach, the Fermi level and thus relative populations of different defect charge states and carrier concentrations re-equilibrate upon cooling (under the constraint of fixed total concentration for each defect), which again is solved self-consistently using the net charge neutrality condition and the constrained defect concentrations.
Warning
In certain cases (such as Li-ion battery materials or extremely slow charge capture/emission), these approximations may have to be adjusted such that some defects/charge states are considered fixed and some are allowed to re-equilibrate (e.g. highly mobile Li vacancies/interstitials). As noted later, the FermiSolver class can be used in these cases for more fine-grained control over constraints and approximations in defect concentration calculations, as demonstrated in the Advanced Defect/Carrier Concentration Analysis tutorial.
import numpy as np
from tqdm import tqdm
anneal_temperatures = np.arange(200, 1401, 50) # annealing temperatures to consider, in K
annealing_dict = {}
for anneal_temp in tqdm(anneal_temperatures):
(fermi_level, e_conc, h_conc, conc_df,
annealing_fermi_level, annealing_e_conc, annealing_h_conc, _annealing_conc_df
) = CdTe_thermo.get_fermi_level_and_concentrations(
bulk_dos=CdTe_thermo.bulk_dos, limit="Cd-rich", annealing_temperature=anneal_temp, skip_formatting=True,
return_annealing_values=True, # return annealing Fermi level & concentrations for comparison
) # quenching to 300K (default) – alternatively can specify quench (operating) temperature
annealing_dict[anneal_temp] = {
"annealing_fermi_level": annealing_fermi_level,
"annealing_e_conc": annealing_e_conc,
"annealing_h_conc": annealing_h_conc,
"fermi_level": fermi_level,
"e_conc": e_conc,
"h_conc": h_conc,
"conc_df": conc_df
}
100%|██████████| 25/25 [00:01<00:00, 22.77it/s]
import matplotlib.pyplot as plt
from doped.utils.plotting import shade_band_edges
f, ax = plt.subplots()
anneal_fermi_levels = np.array([v["annealing_fermi_level"] for k, v in annealing_dict.items()])
quenched_fermi_levels = np.array([v["fermi_level"] for k, v in annealing_dict.items()])
ax.plot(anneal_temperatures, anneal_fermi_levels, marker='o',
label="$E_F$ during annealing (@ $T_{anneal}$)", color = "k", alpha=0.25)
ax.plot(anneal_temperatures, quenched_fermi_levels, marker='o',
label="$E_F$ upon cooling (@ $T$ = 300K)", color = "k", alpha=0.9)
ax.set_xlabel('Anneal Temperature (K)')
ax.set_ylabel('Fermi Level wrt VBM (eV)')
ax.set_xlim(300, 1400)
ax.axvspan(500+273.15, 700+273.15, alpha=0.2, color='#33A7CC', label="Typical Anneal Range")
ax.legend()
# show VB in blue and CB in orange:
ylim = (-0.2, band_gap+0.2)
shade_band_edges(ax, band_gap, ax.get_xlim(), ylim, orientation="vertical")
ax.set_ylim(ylim)
f
print(f"Room-temp Fermi level for T_anneal = {anneal_temperatures[16]} K: {quenched_fermi_levels[16]:.2f} eV above VBM")
Room-temp Fermi level for T_anneal = 1000 K: 1.44 eV above VBM
Let’s plot the defect/carrier concentrations:
annealing_n = np.array([annealing_dict[k]["annealing_e_conc"] for k in anneal_temperatures])
annealing_p = np.array([annealing_dict[k]["annealing_h_conc"] for k in anneal_temperatures])
quenched_n = np.array([annealing_dict[k]["e_conc"] for k in anneal_temperatures])
quenched_p = np.array([annealing_dict[k]["h_conc"] for k in anneal_temperatures])
# let's make some convenience functions for plotting:
def array_from_conc_df(name, annealing_dict, anneal_temperatures):
"""Return a numpy array of the given defect concentration from the annealing_dict."""
return np.array([annealing_dict[temp]["conc_df"].loc[
(name, 0), "Total Concentration (cm^-3)"
] for temp in anneal_temperatures])
def get_significant_defects(conc_df, conc_cutoff=1e12):
"""Return a list of defects with total concentrations above ``conc_cutoff`` in cm^-3"""
defects_concs = zip(
conc_df.index.get_level_values("Defect").unique(),
conc_df["Total Concentration (cm^-3)"].unique(),
)
defects_concs = sorted(defects_concs, key = lambda x: x[1], reverse=True) # sort by concentration
return [defect for defect, conc in defects_concs if conc > 1e12] # cut <10^12 cm^-3
from doped.utils.plotting import format_defect_name
f, ax = plt.subplots()
# plot carrier concentrations:
ax.plot(anneal_temperatures, quenched_p, label='p', linestyle="--", lw=2) # plot p/n (room temp)
ax.plot(anneal_temperatures, quenched_n, label='n', linestyle="--", lw=2)
ax.plot(anneal_temperatures, annealing_p, label='p ($T_{anneal}$)', alpha=0.5, c = "C0", linestyle="--")
ax.plot(anneal_temperatures, annealing_n, label='n ($T_{anneal}$)', alpha=0.5, c = "C1", linestyle="--")
# only plotting defects with significant concentrations here (>10^12 cm^-3 at T=1350K):
conc_df = annealing_dict[1350]["conc_df"]
for defect in get_significant_defects(conc_df):
ax.plot(anneal_temperatures,
array_from_conc_df(defect, annealing_dict, anneal_temperatures),
label=format_defect_name(defect, include_site_info_in_name=True, wout_charge=True), alpha=0.7, linestyle="--", marker="o")
ax.set_xlabel('Anneal Temperature (K)'); ax.set_ylabel(r"Concentration (cm$^{-3}$)")
ax.set_yscale("log") # log scale
ax.set_xlim(300, 1400); ax.set_ylim(1e12, 1e17)
ax.axvspan(500+273.15, 700+273.15, alpha=0.2, color='#33A7CC') # shade in typical anneal range (this is specific to CdTe here!)
ax.legend()
f
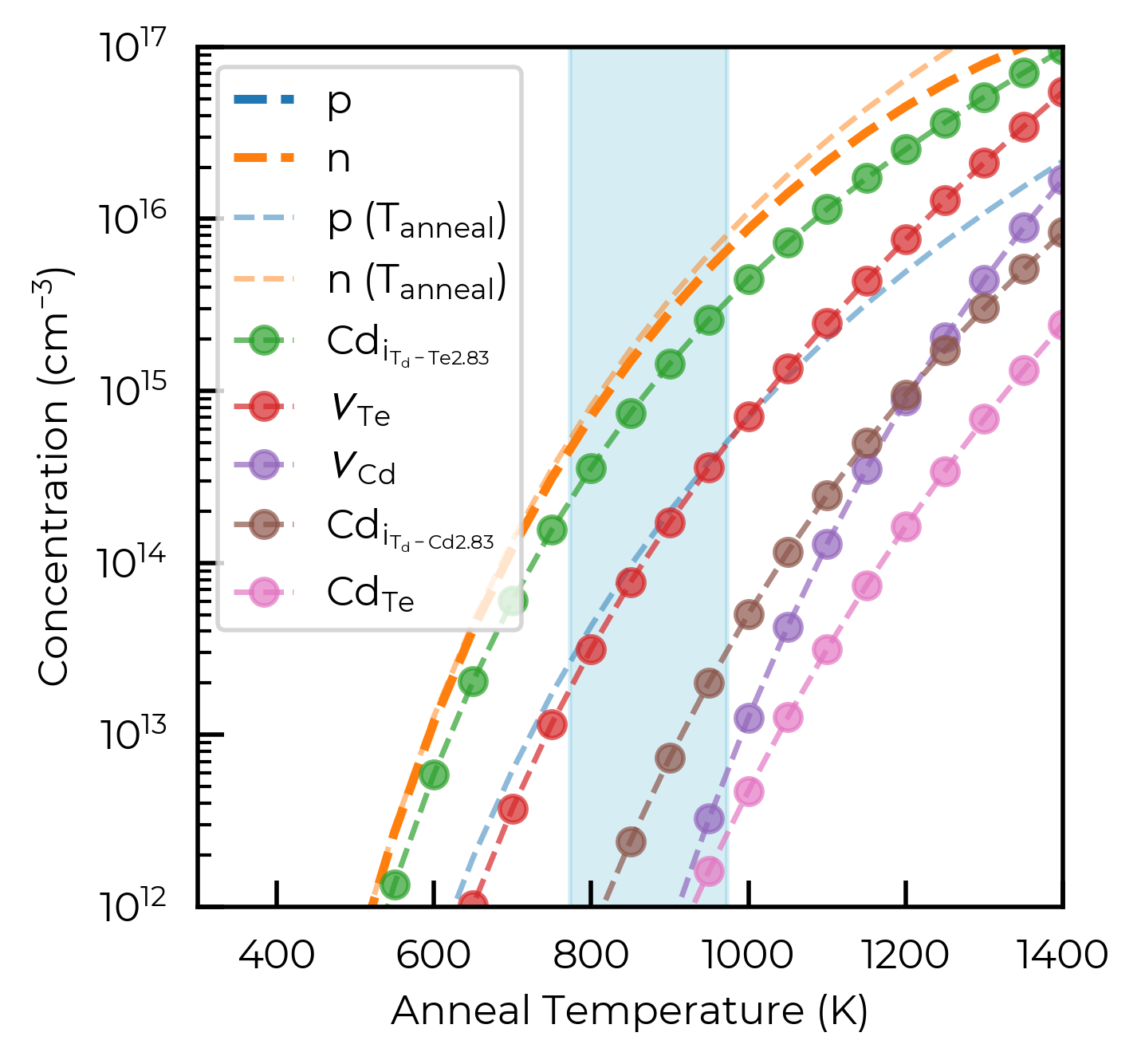
Here we see we get exponential increases in n-type carrier concentration with increasing annealing temperature, as expected, and that the electron concentration primarily originates from \(Cd_{i}^{+2}\) donors (our lowest energy native donor under Cd-rich conditions), with the electron concentration approximately matching the concentration of \(Cd_{i}^{+2}\) (with a factor of 2x) at high temperatures.
Tip
If we had some digitised experimental data then we could also plot together here with our predicted datapoints, as scatter points.
We can of course perform the same analysis for the p-type (Te-rich) limit, or other chemical potential limits / doping conditions. For instance, if we had also calculated the formation energies of extrinsic dopants/impurities, then we could similarly calculate the predicted dopant/defect/carrier concentrations in the presence of these impurities, under various growth conditions.
CdTe: Approximating Temperature-Dependent Band Gap
As with all semiconducting/insulating materials, the band gap (and thus relative energies of defects and charge carriers) in CdTe is dependent on the temperature. These changes in the band edge positions can affect the defect/carrier formation energies & concentrations, and thus the Fermi level position / doping behaviour (discussed in Accurately Modelling Point Defects in Semiconductors: The Case of CdTe, briefly in Sec. III here and exemplified in this paper). In doped, we can account for these changes in band edge positions (if known) using the delta_gap parameter in the get_fermi_level_and_concentrations() method and/or the scissor_dos() function as shown below.
Of course, there are many effects of temperature on the free energies of defect formation (e.g. see Mosquera-Lois et al. Chem Soc Rev 2023, Mosquera-Lois et al. Chem Sci 2025), but here for a crude first approximation we will account for the experimentally-known band gap renormalisation of CdTe as a function of temperature, assuming that the renormalisation occurs symmetrically such that the VBM and CBM eigenvalues are down/up-shifted by the same amount (ΔEg/2) at each temperature while the defect formation energies / transition levels remain fixed.
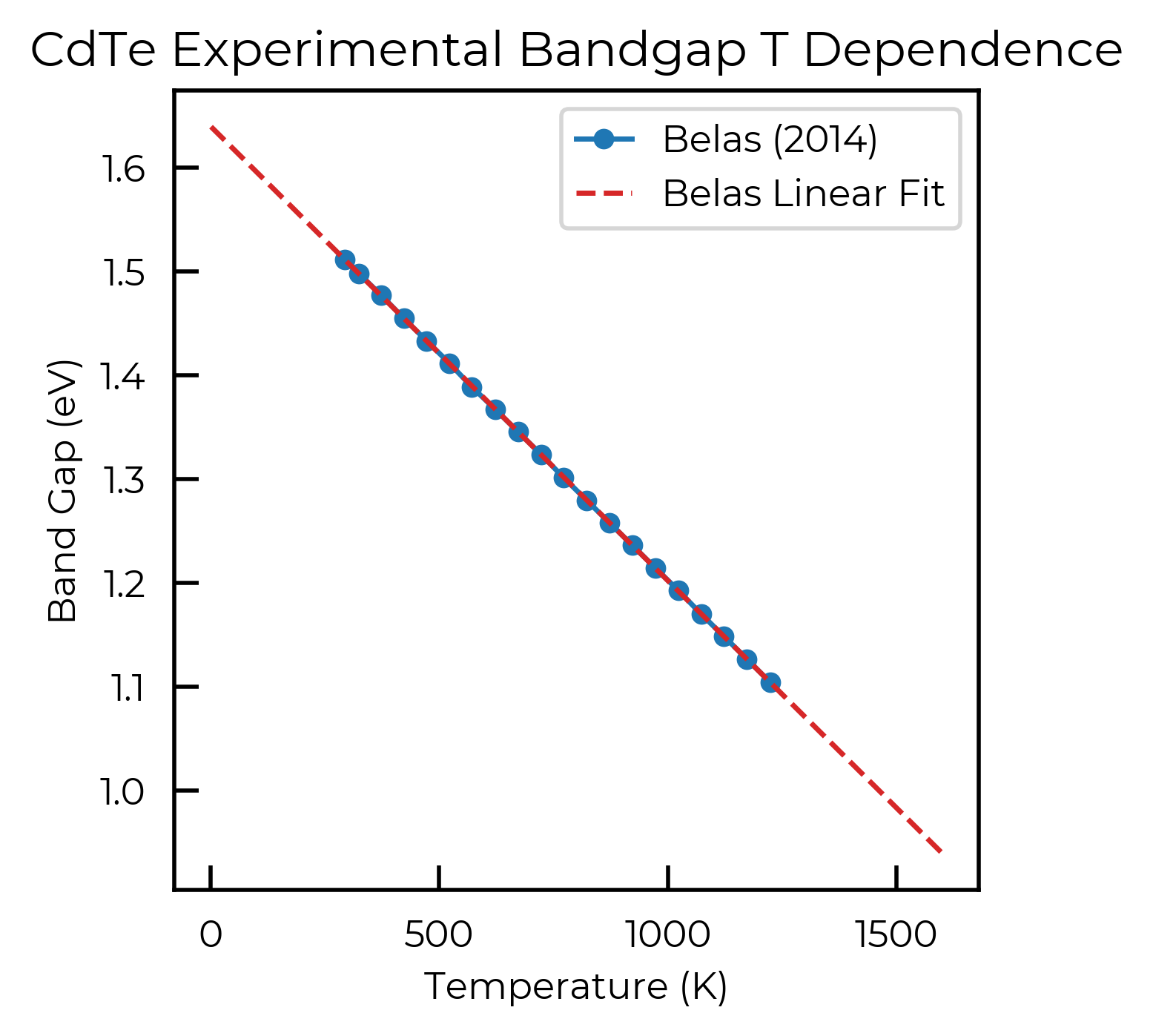
To do this, we first define a function for the temperature-dependent band gap, by fitting to the experimental data:
import numpy as np
band_gap_data = [
(1.5110277123929035, 293.61411498177165), # CdTe gap vs Temp, Belas et al. 2014
(1.4978368122096875, 323.8690412012745),
(1.4771103223451387, 372.54000946743133),
(1.4544939046587835, 422.5264093083491),
(1.4328236569636261, 472.5128091492669),
(1.411153409268469, 522.4992089901847),
(1.3885345794215245, 571.1701772563415),
(1.3668667438869564, 622.4720086720203),
(1.3451964961917993, 672.4584085129379),
(1.3235262484966421, 722.4448083538558),
(1.3009098308102867, 772.4312081947735),
(1.2792395831151295, 822.4176080356914),
(1.2575693354199722, 872.4040078766092),
(1.235899087724815, 922.390407717527),
(1.2142288400296577, 972.3768075584447),
(1.1925585923345006, 1022.3632073993625),
(1.1699421746481453, 1072.3496072402804),
(1.148271926952988, 1122.3360070811982),
(1.1266016792578308, 1172.322406922116),
(1.1039876737320649, 1223.6242383377944),
]
params = np.polyfit([x[1] for x in band_gap_data], [x[0] for x in band_gap_data], 1, full=True)[0]
def band_gap_linear_fit(T): # linear fit of the CdTe experimental band gap vs T
return params[1] + params[0] * T
# alternatively we could also use a Cubic Spline fit such as this (or any other fitted function):
# from scipy.interpolate import CubicSpline
# band_gap_spline_fit = CubicSpline([x[1] for x in band_gap_data], [x[0] for x in band_gap_data])
Let’s quickly plot the experimental data and our linear fit, to confirm it looks reasonable:
f, ax = plt.subplots()
temps = np.linspace(1, 1600, num=1000)
ax.plot([x[1] for x in band_gap_data], [x[0] for x in band_gap_data],
label = "Belas (2014)", marker="o", markersize=4)
ax.plot(temps, band_gap_linear_fit(temps), color="C3", ls="--", label="Belas Linear Fit")
ax.set_xlabel("Temperature (K)"); ax.set_ylabel("Band Gap (eV)")
ax.set_title("CdTe Experimental Bandgap T Dependence")
ax.legend()
f
Now we use this function with the delta_VBM and delta_CBM parameters in get_fermi_level_and_concentrations() to account for the temperature-dependent band gap shift (here applied symmetrically about the original gap):
CdTe: Te-Rich with Temperature Dependence
anneal_temperatures = np.arange(200, 1401, 50) # annealing temperatures to consider, in K
annealing_dict = {}
for anneal_temp in tqdm(anneal_temperatures):
band_gap_shift = band_gap_linear_fit(anneal_temp) - 1.5 # 1.5 eV is our DFT (and room temp) gap
(fermi_level, e_conc, h_conc, conc_df,
annealing_fermi_level, annealing_e_conc, annealing_h_conc, _annealing_conc_df
) = CdTe_thermo.get_fermi_level_and_concentrations(
bulk_dos=CdTe_thermo.bulk_dos, limit="Te-rich", annealing_temperature=anneal_temp, skip_formatting=True,
delta_VBM=-band_gap_shift/2,
delta_CBM=band_gap_shift/2,
return_annealing_values=True, # return annealing Fermi level & concentrations for comparison
) # We use `delta_VBM` and `delta_CBM` (positive = upshift) to specify band-edge shifts
# from the original DFT DOS to the anneal temp; here applied symmetrically (Eg/2 either way).
# See the ZGO section at the end of this tutorial for the asymmetric case.
annealing_dict[anneal_temp] = {
"annealing_fermi_level": annealing_fermi_level,
"annealing_e_conc": annealing_e_conc,
"annealing_h_conc": annealing_h_conc,
"fermi_level": fermi_level,
"e_conc": e_conc,
"h_conc": h_conc,
"conc_df": conc_df
}
100%|██████████| 25/25 [00:01<00:00, 23.75it/s]
Plot the Fermi level (during annealing & upon cooling), and the simulated band edges:
import matplotlib.pyplot as plt
from doped.utils.plotting import shade_band_edges
f, ax = plt.subplots()
anneal_fermi_levels = np.array([v["annealing_fermi_level"] for k, v in annealing_dict.items()])
quenched_fermi_levels = np.array([v["fermi_level"] for k, v in annealing_dict.items()])
ax.plot(anneal_temperatures, anneal_fermi_levels, marker='o',
label="$E_F$ during annealing (@ $T_{anneal}$)", color = "k", alpha=0.25)
ax.plot(anneal_temperatures, quenched_fermi_levels, marker='o',
label="$E_F$ upon cooling (@ $T$ = 300K)", color = "k", alpha=0.9)
ax.set_xlabel('Anneal Temperature (K)')
ax.set_ylabel('Fermi Level wrt VBM (eV)')
ax.set_xlim(300, 1400)
ax.axvspan(500+273.15, 700+273.15, alpha=0.2, color='#33A7CC', label="Typical Anneal Range")
ax.fill_between(anneal_temperatures, (1.5 - band_gap_linear_fit(anneal_temperatures))/2,
0, alpha=0.2, color="C0", label="VBM (T @ $T_{anneal}$)", linewidth=0.25,)
ax.fill_between(anneal_temperatures, 1.5 - (1.5 - band_gap_linear_fit(anneal_temperatures))/2,
1.5, alpha=0.2, color="C1", label="CBM (T @ $T_{anneal}$)", linewidth=0.25,)
ax.legend(loc="upper center")
# show VB in blue and CB in orange:
ylim = (-0.2, band_gap+0.2)
shade_band_edges(ax, band_gap, ax.get_xlim(), ylim, orientation="vertical")
ax.set_ylim(ylim)
f
print(f"Room-temp Fermi level for T_anneal = {anneal_temperatures[16]} K: {quenched_fermi_levels[16]:.2f} eV above VBM")
Room-temp Fermi level for T_anneal = 1000 K: 0.32 eV above VBM
Let’s plot the defect/carrier concentrations:
annealing_n = np.array([annealing_dict[k]["annealing_e_conc"] for k in anneal_temperatures])
annealing_p = np.array([annealing_dict[k]["annealing_h_conc"] for k in anneal_temperatures])
quenched_n = np.array([annealing_dict[k]["e_conc"] for k in anneal_temperatures])
quenched_p = np.array([annealing_dict[k]["h_conc"] for k in anneal_temperatures])
from doped.utils.plotting import format_defect_name
f, ax = plt.subplots()
# plot carrier concentrations:
ax.plot(anneal_temperatures, quenched_p, label='p', linestyle="--", lw=2) # plot p/n (room temp)
ax.plot(anneal_temperatures, quenched_n, label='n', linestyle="--", lw=2)
ax.plot(anneal_temperatures, annealing_p, label='p ($T_{anneal}$)', alpha=0.5, c = "C0", linestyle="--")
ax.plot(anneal_temperatures, annealing_n, label='n ($T_{anneal}$)', alpha=0.5, c = "C1", linestyle="--")
# only plotting defects with significant concentrations here (>10^12 cm^-3 at T=1350K):
conc_df = annealing_dict[1350]["conc_df"]
for defect in get_significant_defects(conc_df):
ax.plot(anneal_temperatures,
array_from_conc_df(defect, annealing_dict, anneal_temperatures),
label=format_defect_name(defect, include_site_info_in_name=True, wout_charge=True), alpha=0.7, linestyle="--", marker="o")
ax.set_xlabel('Anneal Temperature (K)'); ax.set_ylabel(r"Concentration (cm$^{-3}$)")
ax.set_yscale("log") # log scale
ax.set_xlim(300, 1400); ax.set_ylim(1e12, 1e18)
ax.axvspan(500+273.15, 700+273.15, alpha=0.2, color='#33A7CC') # shade in typical anneal range (this is specific to CdTe here!)
ax.legend()
f

Let’s compare to some experimental data of p at T-anneal:
wienecke_data = np.array(
[ # Wienecke et al. 1993
[675.644735186816, 15.19509584755584],
[774.64775443452, 15.983458618047331],
[773.2859479179771, 15.780402388747808],
[876.594540193735, 16.456749859094277],
[866.7316643602969, 16.470175483037483],
[931.3592904767895, 16.68944378653258],
[972.2040508240029, 16.939464368267398],
[1043.955214492389, 17.234473455894925],
[1030.0320795068562, 17.11399747952909],
[1077.6449867907913, 17.335494943226077],
[1082.4820732167568, 17.165318826904443],
]
)
emanuelsson_data = np.array([[750 + 273.15, np.log10(1.2e17)]]) # Emanuelsson et al. 1993
expt_data = np.append(wienecke_data, emanuelsson_data, axis=0)
from doped.utils.plotting import format_defect_name
f, ax = plt.subplots()
# plot carrier concentrations:
ax.plot(anneal_temperatures, quenched_p, label='p', linestyle="--", lw=2) # plot p/n (room temp)
ax.plot(anneal_temperatures, quenched_n, label='n', linestyle="--", lw=2)
ax.plot(anneal_temperatures, annealing_p, label='p ($T_{anneal}$)', alpha=0.5, c = "C0", linestyle="--")
ax.plot(anneal_temperatures, annealing_n, label='n ($T_{anneal}$)', alpha=0.5, c = "C1", linestyle="--")
# only plotting defects with significant concentrations here (>10^12 cm^-3 at T=1350K):
conc_df = annealing_dict[1350]["conc_df"]
for defect in get_significant_defects(conc_df):
ax.plot(anneal_temperatures,
array_from_conc_df(defect, annealing_dict, anneal_temperatures),
label=format_defect_name(defect, include_site_info_in_name=True, wout_charge=True), alpha=0.7, linestyle="--", marker="o")
ax.scatter(expt_data[:,0], 10**(expt_data[:,1]), marker="x", label="Expt", c="C0",
alpha=0.5)
ax.set_xlabel('Anneal Temperature (K)'); ax.set_ylabel(r"Concentration (cm$^{-3}$)")
ax.set_yscale("log") # log scale
ax.set_xlim(300, 1400); ax.set_ylim(1e12, 1e18)
ax.axvspan(500+273.15, 700+273.15, alpha=0.2, color='#33A7CC') # shade in typical anneal range (this is specific to CdTe here!)
ax.legend()
f

Here there is almost certainly some fortuitous error cancellation at play, but we see we get a reasonable qualitative agreement with the experimental data.
Note
The same fitted-bandgap-function approach used here can be applied with calculated data (e.g. from DFT electron-phonon coupling calculations), and can additionally account for asymmetric band-edge renormalisation using the delta_VBM and delta_CBM parameters (positive = upshift), as demonstrated in the ZGO example below.
Carrier / Defect concentrations vs (μ, T)
To analyse the behaviour of native defects & carrier concentrations in CdTe more extensively, let’s scan over a grid of chemical potentials and annealing temperatures, and calculate the corresponding defect and carrier concentrations. To do this, let’s first define a function which interpolates over our chemical potential space in one-dimension:
from doped.chemical_potentials import get_X_rich_poor_limit
# here we're going from Te-poor to Te-rich, but this can be adjusted for your specific case!
poor_limit_chempots = CdTe_thermo.chempots[
"limits_wrt_el_refs"][get_X_rich_poor_limit("Te-poor", CdTe_thermo.chempots)] # get the Te-poor limit
rich_limit_chempots = CdTe_thermo.chempots[
"limits_wrt_el_refs"][get_X_rich_poor_limit("Te-rich", CdTe_thermo.chempots)] # get the Te-rich limit
def chempots_at_x(x, poor_limit_chempots=poor_limit_chempots, rich_limit_chempots=rich_limit_chempots):
x_dict = {}
for el, poor_val in poor_limit_chempots.items():
x_dict[el] = poor_val + x * (rich_limit_chempots[el] - poor_val)
return x_dict
Tip
For a binary system like CdTe, the choice of chemical potential points to interpolate between is simple, but for more complex ternary/quaternary etc systems, you may want to test a few chemical potential limits to interpolate between!
We can then run this loop to calculate our defect/carrier concentrations over a range of temperatures and chemical potentials, and save the data to file to prevent having to re-run this parsing:
Tip
This loop takes ~2 minutes to run on a Macbook Pro, but if you want to skip this, you can just load the pre-computed data with dict_list = loadfn("CdTe/CdTe_2D_defect_carrier_concentrations.json.gz") as shown in the next cell.
from joblib import Parallel, cpu_count, delayed
from monty.serialization import dumpfn
from tqdm import tqdm
anneal_temperatures = np.linspace(300, 1400, 100) # 100 temperature datapoints from 300K to 1400K
chempot_x = np.linspace(0, 1, 100) # 100 chemical potential datapoints from X-poor to X-rich
kwargs_list = []
for x in chempot_x:
relative_chempots = chempots_at_x(x)
for anneal_temp in anneal_temperatures:
band_gap_shift = band_gap_linear_fit(anneal_temp) - 1.5 # 1.5 eV is our DFT (and room temp) gap
# We use `delta_VBM` and `delta_CBM` (positive = upshift) to specify band-edge shifts
# from the original DFT DOS to the anneal temp; here applied symmetrically. See the
# ZGO section at the end of the tutorial for the asymmetric case.
kwargs_list.append(
{
"annealing_temperature": anneal_temp,
"delta_VBM": -band_gap_shift / 2,
"delta_CBM": band_gap_shift / 2,
"chempots": relative_chempots,
"skip_formatting": True,
"return_annealing_values": True,
}
)
# here I use joblib Parallel multiprocessing to speed up computation over this large grid
# with batch_size set flexibly to optimise speed
results = Parallel(n_jobs=-1, verbose=1, batch_size=int(len(kwargs_list) / (cpu_count() * 10)))(
delayed(CdTe_thermo.get_fermi_level_and_concentrations)(**kwargs) for kwargs in tqdm(kwargs_list)
) # 3.5 mins on MacBook pro
# using the built-in multiprocessing module is awkward here as it doesn't allow kwargs to be
# passed to the underlying function, or for picklable functions to be defined in a notebook
# alternatively we can run without parallel processing, with:
# results = [
# CdTe_thermo.get_fermi_level_and_concentrations(**kwargs) for kwargs in tqdm(kwargs_list)]
0%| | 0/10000 [00:00<?, ?it/s][Parallel(n_jobs=-1)]: Using backend LokyBackend with 10 concurrent workers.
30%|███ | 3020/10000 [00:46<01:39, 69.85it/s][Parallel(n_jobs=-1)]: Done 3020 tasks | elapsed: 56.8s
100%|██████████| 10000/10000 [02:08<00:00, 77.98it/s]
[Parallel(n_jobs=-1)]: Done 9830 tasks | elapsed: 2.6min
[Parallel(n_jobs=-1)]: Done 10000 out of 10000 | elapsed: 2.7min finished
dict_list = []
for kwargs, result in zip(kwargs_list, results):
(fermi_level, e_conc, h_conc, conc_df,
annealing_fermi_level, annealing_e_conc, annealing_h_conc, _annealing_conc_df
) = result
conc_dict = {
"annealing_fermi_level": annealing_fermi_level,
"annealing_e_conc": annealing_e_conc,
"annealing_h_conc": annealing_h_conc,
"fermi_level": fermi_level,
"e_conc": e_conc,
"h_conc": h_conc,
"conc_df": conc_df,
}
dict_list.append((kwargs.get("chempots"), kwargs.get("annealing_temperature"), conc_dict))
dumpfn(dict_list, "CdTe/CdTe_2D_defect_carrier_concentrations.json.gz")
# # if skipping re-calculation above, run this:
# from monty.serialization import loadfn
# dict_list = loadfn("CdTe/CdTe_2D_defect_carrier_concentrations.json.gz")
Plot carrier concentrations as a 2D heatmap:
import matplotlib
from matplotlib.colors import SymLogNorm
# get doping concentrations (p - n) to plot:
dict_array_vals = {k: np.array([conc_dict[k] for conc_dict in np.array(dict_list)[:, 2]]) for k in dict_list[0][2].keys()}
doping_concentration = dict_array_vals["h_conc"] - dict_array_vals["e_conc"] # p - n
f, ax = plt.subplots()
# define diverging colourmap, from blue to white to orange, quadratically:
colour_list = ["C0", "#D8E6F1", "white", "#FDE5D0", "C1"]; colour_list.reverse() # of course can use any other colourmap of your choosing here
cmap = matplotlib.colors.LinearSegmentedColormap.from_list("", colour_list)
mappable = ax.imshow(
doping_concentration.reshape(len(chempot_x), len(anneal_temperatures)),
extent=[min(anneal_temperatures), max(anneal_temperatures), min(chempot_x), max(chempot_x)], # reverse y axis
aspect="auto", origin="lower", cmap=cmap, interpolation="gaussian",
norm=SymLogNorm(linthresh=5e11, vmin=-1e17, vmax=1e17)) # set normalisation of colourmap
# add contour lines at p = 1e13, n = 1e13, 1e14, 1e15, 1e16 to help illustrate:
ax.contour(anneal_temperatures, chempot_x,
doping_concentration.reshape(len(chempot_x), len(anneal_temperatures)),
levels=[-1e16, -1e15, -1e14, -1e13], colors="k", alpha=0.5, linestyles="--")
ax.contour(anneal_temperatures, chempot_x,
doping_concentration.reshape(len(chempot_x), len(anneal_temperatures)),
levels=[1e13, ], colors="k", alpha=0.5, linestyles="--")
ax.set_xlim(500, 1400); ax.set_yticks([0, 1])
ax.set_yticklabels(["Cd-Rich", "Te-Rich"]) # here Te-poor = Cd-rich
cbar = f.colorbar(mappable, ax=ax, ticks=[-1e17, -1e15, -1e13, 0, 1e13, 1e15, 1e17])
cbar.set_label(r"$p\ -\ n$ (cm$^{-3}$)") # add and label colourbar
ax.set_xlabel("Anneal Temperature (K)"); ax.set_ylabel("Chemical Potential", labelpad=-30)
f

Here we see that while a greater portion of chemical potential & temperature space for intrinsic CdTe results in p-type doping, the hole concentration remains relatively low, just above \(10^{13}\) cm\(^{-3}\). Under Cd-rich conditions on the other hand, we obtain much higher electron carrier concentrations (~\(10^{17}\) cm\(^{-3}\) at high temperatures).
We can perform similar heatmap analyses of specific defect concentrations, Fermi level positions etc.
CdTe: Defect Concentrations vs μ
Finally, let’s plot the defect concentrations as a function of chemical potential, at a fixed annealing temperature of 900 K:
dict_list_900K = [d for d in dict_list if d[1] == 900] # select dicts for 900K annealing
dict_array_vals_900K = {
k: [conc_dict[k] for conc_dict in np.array(dict_list_900K)[:, 2]] for k in dict_list_900K[0][2].keys()
}
conc_df_array_900K = dict_array_vals_900K["conc_df"]
defect_conc_dict_900K = {
defect_charge_tuple[0]: np.array([
conc_df_array_900K[i]["Total Concentration (cm^-3)"].loc[defect_charge_tuple]
for i in range(len(conc_df_array_900K))]) for defect_charge_tuple in conc_df_array_900K[0].index
}
f, ax = plt.subplots(figsize=(7, 3.5))
plt.rcParams["lines.markersize"] = 4
# plot carrier concentrations:
ax.plot(chempot_x, dict_array_vals_900K["h_conc"], label='p', lw=2, linestyle="--")
ax.plot(chempot_x, dict_array_vals_900K["e_conc"], label='n', lw=2, linestyle="--")
ax.plot(chempot_x, dict_array_vals_900K["annealing_h_conc"],
label='p ($T_{anneal}$)', c="C0", linestyle="--", alpha=0.5)
ax.plot(chempot_x, dict_array_vals_900K["annealing_e_conc"],
label='n ($T_{anneal}$)', c="C1", linestyle="--", alpha=0.5)
for defect, conc_array in defect_conc_dict_900K.items():
ax.plot(chempot_x, conc_array,
label=format_defect_name(defect, include_site_info_in_name=True, wout_charge=True), alpha=0.7, linestyle="--", marker="o")
ax.set_xlabel("Chemical Potential"); ax.set_ylabel(r"Concentration (cm$^{-3}$)")
ax.set_yscale("log"); ax.set_xlim(0, 1); ax.set_ylim(8e11, 1.35e16)
ax.set_xticks([0,1]); ax.set_xticklabels(["Cd-Rich", "Te-Rich"]) # here Te-poor = Cd-rich
# add ticks and ticklabels on both left and right yaxis:
ax3 = ax.twinx(); ax3.set_ylim(8e11, 1.35e16); ax3.set_yscale("log")
f.legend(loc="right", bbox_to_anchor=(1.15, 0.5))
f.tight_layout()
f

Tip
Note that the effects of metastable defect states on defect & carrier concentrations are included in this analysis, where they may have significant concentrations particularly at elevated annealing temperatures, contributing to the ‘effective degeneracy’ and thus overall concentration of the defect species.
from doped.utils.plotting import shade_band_edges
f, ax = plt.subplots(figsize=(7, 3.5))
ax.plot(chempot_x, dict_array_vals_900K["annealing_fermi_level"], marker='o',
label="$E_F$ during annealing (@ $T_{anneal}$)", color = "k", alpha=0.25)
ax.plot(chempot_x, dict_array_vals_900K["fermi_level"], marker='o',
label="$E_F$ upon cooling (@ $T$ = 300K)", color = "k", alpha=0.9)
ax.set_xlabel("Chemical Potential"); ax.set_ylabel('Fermi Level wrt VBM (eV)')
ax.set_xlim(0, 1); ax.set_xticks([0, 1])
ax.set_xticklabels(["Cd-Rich", "Te-Rich"]) # here Te-poor = Cd-rich
ax3 = ax.twinx(); ax3.set_ylim(-0.2, band_gap+0.2) # show tick labels on both sides
ax.set_ylim(-0.2, band_gap+0.2); ax.legend()
# show VB in blue and CB in orange:
ylim = (-0.2, band_gap + 0.2)
shade_band_edges(ax, band_gap, ax.get_xlim(), ylim, orientation="vertical")
ax.set_ylim(ylim)
f
ZGO: Asymmetric Band-Edge Renormalisation
In the above example with CdTe, we assumed that temperature-induced band gap renormalisation occurred symmetrically, such that the VBM/CBM shifted upwards/downwards by \(-\Delta E_g\)/2. This is often a (surprisingly) reasonable assumption (shown for instance in Dopability limits in Al-rich AlGaN alloys for far-UVC LEDs – Ling et al. arXiv 2026, and above / in this thesis for CdTe) – particularly when the dispersion / effective masses of the VBM and CBM are comparable – but of course a more accurate treatment should incoporate the individual temperature dependences of the band edges (or even more accurately, the full temperature-dependent electronic density of states) when the data is available, as supported through the delta_VBM and delta_CBM parameters for DefectThermodynamics.get_fermi_level_and_concentrations (and the FermiSolver methods).
An example of asymmetric, temperature-dependent band-edge renormalisation is given by ZnGa₂O₄, investigated by Claes et al. ACS Appl. Mater. Interfaces 2025, where band-edge temperature dependence was directly computed using DFPT electron-phonon calculations. Here we use this data to exemplify the delta_VBM and delta_CBM parameters and effects of asymetric band-edge renormalisation.
import numpy as np
from monty.serialization import loadfn
ZGO_thermo = loadfn("ZGO/ZGO_thermo.json.gz")
ZGO_thermo.bulk_dos = "ZGO/ZGO_bulk_DOS_vasprun.xml.gz"
# Linear fits to DFPT-derived VBM(T) and CBM(T) shifts in ZGO (eV, T in K) from Claes et al.
# the VBM shifts up while the CBM shifts down (gap shrinks asymmetrically with T)
def VBM_shift(T):
return 0.1662 + 3.01e-4 * T
def CBM_shift(T):
return -0.0756 - 2.04e-4 * T
from tqdm import tqdm
anneal_temperatures = np.arange(300, 2101, 50) # processing/annealing temperatures (K)
results = {"none": {}, "asymmetric": {}, "symmetric": {}}
common_kwargs = {
"quenched_temperature": 300,
"limit": "Zn(GaO2)2-Zn-Ga",
"skip_formatting": True,
"return_annealing_values": True,
}
for T in tqdm(anneal_temperatures):
dv = VBM_shift(T)
dc = CBM_shift(T)
delta_gap = dc - dv # change in gap (sym. equivalent)
results["none"][T] = ZGO_thermo.get_fermi_level_and_concentrations(
annealing_temperature=T,
**common_kwargs,
)
results["asymmetric"][T] = ZGO_thermo.get_fermi_level_and_concentrations(
annealing_temperature=T,
delta_VBM=dv,
delta_CBM=dc,
**common_kwargs,
)
results["symmetric"][T] = ZGO_thermo.get_fermi_level_and_concentrations(
annealing_temperature=T,
delta_VBM=-delta_gap / 2,
delta_CBM=+delta_gap / 2,
**common_kwargs,
)
0%| | 0/37 [00:00<?, ?it/s]The defect supercell has been detected to possibly have a non-scalar matrix expansion, which could be breaking the cell periodicity and possibly preventing the correct _relaxed_ point group symmetries (and thus orientational degeneracies) from being automatically determined.
This will not affect defect formation energies / transition levels, but can be important for concentrations/doping/Fermi level behaviour (10.1039/D2FD00043A, 10.1039/D3CS00432E, 10.1038/s41578-025-00879-y ...).
You can manually check (and edit) the computed defect point symmetries and corresponding orientational degeneracy factors by inspecting/editing the calculation_metadata['relaxed point symmetry'] and degeneracy_factors['orientational degeneracy'] attributes.
100%|██████████| 37/37 [00:03<00:00, 10.04it/s]
Below we compare the predicted (quenched / room-temperature) electron concentrations for three band-edge renormalisation scenarios (none, symmetric and asymmetric renormalisation – left), and show the corresponding Fermi level evolution and asymmetric temperature-dependent VBM/CBM shifts for the assymetric (highest-fidelity) case (right).
Note
Here for illustration we compute three example scenarios; with no band-edge temperature effects, with symmetric band-edge renormalisation and with full asymmetric band-edge renormalisation. In an actual research investigation of course, you would likely just use the highest fidelity data you have available!
import matplotlib.pyplot as plt
import doped
from doped.utils.plotting import shade_band_edges
plt.style.use(f"{doped.__path__[0]}/utils/doped.mplstyle") # use doped style
band_gap = ZGO_thermo.band_gap
f, axes = plt.subplots(1, 2, figsize=(7, 2.75), constrained_layout=True)
# Electron concentration comparison across renormalization cases:
axes[0].axhspan(7.5e19, 9.5e19, color="teal", alpha=0.25, label="Max exp. n concentrations")
axes[0].semilogy(
anneal_temperatures,
[results["symmetric"][T][1] for T in anneal_temperatures],
ls="--",
lw=2.5,
color="C2",
label="With symmetric T-dep bandgap",
)
axes[0].semilogy(
anneal_temperatures,
[results["asymmetric"][T][1] for T in anneal_temperatures],
ls="--",
lw=2.5,
color="C0",
label="With T-dep bandgap",
)
axes[0].semilogy(
anneal_temperatures,
[results["none"][T][1] for T in anneal_temperatures],
ls="--",
lw=2.5,
color="C1",
label="Without T-dep bandgap",
)
axes[0].set_xlabel("Processing Temperature (K)")
axes[0].set_ylabel("Electron Concentration (cm$^{-3}$)")
axes[0].set_xlim(300, 2100)
axes[0].set_ylim(1e15, 1e21)
axes[0].legend()
# Fermi level and asymmetric band-edge renormalisation:
anneal_fermi = np.array([results["asymmetric"][T][4] for T in anneal_temperatures])
quenched_fermi = np.array([results["asymmetric"][T][0] for T in anneal_temperatures])
vbm_T = np.array([VBM_shift(T) for T in anneal_temperatures])
cbm_T = band_gap + np.array([CBM_shift(T) for T in anneal_temperatures])
axes[1].plot(
anneal_temperatures,
anneal_fermi,
marker="o",
label=r"$E_F$ (@ $T_{\mathrm{anneal}}$)",
color="k",
alpha=0.25,
)
axes[1].plot(
anneal_temperatures,
quenched_fermi,
marker="o",
label=r"$E_F$ (@ 300 K)",
color="k",
alpha=0.9,
)
axes[1].fill_between(
anneal_temperatures,
vbm_T,
0,
alpha=0.2,
color="C0",
label=r"VBM ($T_{\mathrm{anneal}}$)",
linewidth=0.25,
)
axes[1].fill_between(
anneal_temperatures,
cbm_T,
band_gap,
alpha=0.2,
color="C1",
label=r"CBM ($T_{\mathrm{anneal}}$)",
linewidth=0.25,
)
axes[1].set_xlabel("Processing Temperature (K)")
axes[1].set_ylabel("Fermi Level wrt VBM (eV)")
axes[1].set_xlim(300, 2100)
axes[1].legend()
# show VB in blue and CB in orange:
ylim = (
min(vbm_T.min(), quenched_fermi.min(), anneal_fermi.min(), -0.2) - 0.05,
max(cbm_T.max(), quenched_fermi.max(), anneal_fermi.max(), band_gap + 0.2) + 0.05,
)
shade_band_edges(axes[1], band_gap, axes[1].get_xlim(), ylim, orientation="vertical")
ax.set_ylim(ylim)
plt.show()
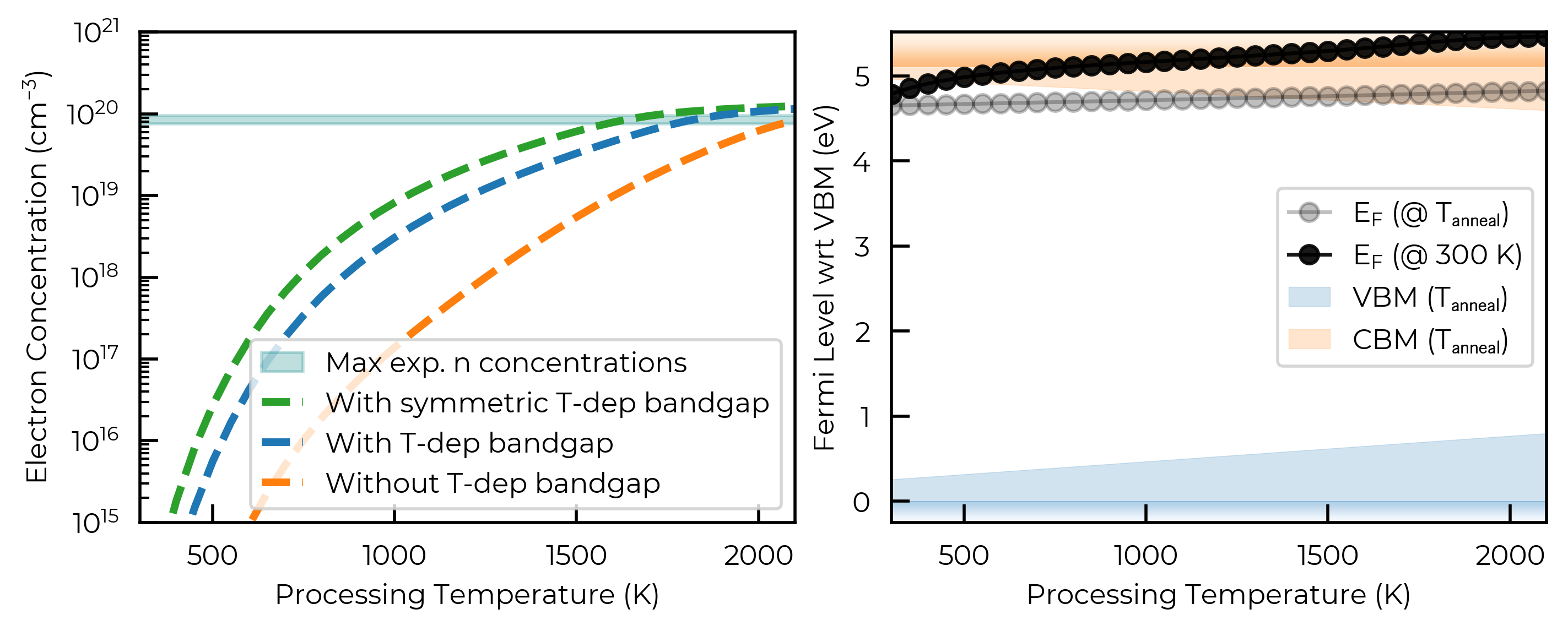
Of course, further temperature effects such as vibrational entropy can be important for quantitatively-accurate defect predictions, for which we direct the reader to 10.1039/D3CS00432E, 10.1039/D4SC08582E, 10.1039/D4CP04817B.
Notes
Important
We can perform all this analysis when we have included extrinsic defects in our calculated defect dataset as well, in order to investigate the predicted doping behaviour of extrinsic species.
Tip
The ’frozen defect’ approximation illustrated in these examples may not be totally applicable in cases where the material is slowly cooled from the annealing temperatures (allowing quasi-equilibrium to be attained at lower temperatures) or for highly-mobile defects which can migrate and potentially annihilate at room/operating temperatures (e.g. Li in battery materials).
In such cases, more detailed/complex models are required to accurately predict the defect and carrier thermodynamics and electronic response. The FermiSolver class in doped.thermodynamics can be used to implement these more fine-grained constraints and approximations in defect concentration calculations; such as specifying (known) fixed concentrations of a defect/dopant, specifying defects to exclude from high-temperature concentration fixing in the frozen defect approximation (e.g. highly-mobile defects), and/or fixing defect charge states upon quenching – as well as implementing a number of convenience methods for thermodynamic analyses; such as scanning over temperatures, chemical potentials, effective dopant concentrations etc, minimising or maximising a target property (e.g. defect/carrier concentration). See the Advanced Defect/Carrier Concentration Analysis for example usage.