Tips & Tricks
The development philosophy behind doped has been to build a powerful, efficient and flexible code
for managing and analysing solid-state defect calculations, having reasonable defaults (that work well for
the majority of materials/defects) with flexibility for the user to customise the workflow to
their specific needs/system.
Note
While we provide some general rules-of-thumb for reasonable choices in the calculation workflow
(based on the literature and our experience), there is no substitute for the user’s own judgement.
Defect behaviour is system-dependent, so it is always important to question and
consider the choices and approximations made in the workflow (such as supercell choice, charge state
ranges, interstitial site pruning, MAGMOM initialisation etc.) in the context of your specific
host system.
See the Literature section on the tutorials page for recommended literature on defect calculations, in particular we strongly recommend Guidelines for robust and reproducible point defect simulations in crystals which addresses many of the common pitfalls and best practices for defect simulations.
Interstitials
Voronoi tessellation is used by default to generate the candidate interstitial sites in doped. We have
consistently found this approach to be the most robust in identifying all stable/low-energy interstitial
sites across a wide variety of materials and chemistries, when combined with structure-searching approaches
such as ShakeNBreak. A nice discussion is given in
Kononov et al. J. Phys.: Condens. Matter 2023.

Workflow for interstitial site generation with Voronoi tessellation defaults in doped, adapted from
JPhys Energy 2025.
As with all aspects of the calculation workflow, interstitial site generation is flexible, and you can
explicitly specify the interstitial sites to generate using the interstitial_coords (for instance, if
you only want to investigate one specific known interstitial site, or input a list of candidate sites
generated from a different algorithm), and/or customise the generation algorithm via
interstitial_gen_kwargs, both of which are input parameters for the
DefectsGenerator class.
Note
Charge-density based approaches for interstitial site generation can be useful in some cases and often output fewer candidate sites, but we have found that these are primarily suited to ionic materials (and with fully-ionised defect charge states) where electrostatics primarily govern the energetics. In many systems (particularly those with some presence of (ionic-)covalent bonding) where orbital hybridisation plays a role, this approach can often miss the ground-state interstitial site(s).
Efficient Interstitial Screening
As described in the defect calculation tutorial (YouTube,
B站),
our recommended workflow for calculating interstitial defects is to first generate the set of candidate
interstitial sites for your structure using DefectsGenerator (which uses Voronoi
tessellation for this, see note below), and then perform Gamma-point-only relaxations (using vasp_gam)
for each charge state of the generated interstitial candidates, and then pruning some of the candidate
sites based on the criteria below. Typically the easiest way to do this is to follow the workflow shown in
the defect generation tutorial, and then run the ShakeNBreak vasp_gam relaxations for the
Unperturbed and Bond_Distortion_0.0%/Rattled directories of each charge state. Alternatively,
you can generate the vasp_gam relaxation input files by setting vasp_gam = True in
DefectsSet write_files() – this will rattle
the output structures by default to break symmetry (controlled by the rattle option).
We can then compare the energies of these trial relaxations, and remove candidates that either:
Are high energy (~>1 eV above the lowest energy site for each charge state), and so are unlikely to form.
Relax to the same final structure/energy as other interstitial sites (despite different initial positions) in each charge state, and so are unnecessary to calculate. This can happen due to interstitial migration within the relaxation calculation, from an unfavourable higher energy site, to a lower energy one. Typically if the energy from the test
vasp_gamrelaxations are within a couple of meV of each other, this is the case.
See the workflow summary on DeepWiki.
Tip
As with many steps in the defect calculation workflow, these are only rough general guidelines and you should always critically consider the validity of these choices in the context of your specific system (for example, considering the charge-state dependence of the interstitial site formation energies here).
Tip
If computational resources are limited, this screening step can often be accelerated without significantly compromising accuracy by just considering the fully-ionised and neutral charge states of each interstitial site, and pruning based on those results.
Performance Bottlenecks
Generation
For complex, low-symmetry systems, defect generation can take a little time (on the order of a couple
minutes). The doped algorithms have been heavily-optimised to expedite this process, and will use
multiprocessing by default to accelerate when multiple CPUs are available. The routines which are typically
the most computationally expensive for generation are supercell generation, interstitial generation and
Wyckoff/symmetry analysis (in order). As such, if you want to accelerate the generation process, you can
expedite these steps by:
Providing your known desired supercell as input and setting
generate_supercell=False, to skip supercell generation (or using tighter supercell generation constraints withsupercell_gen_kwargs).Skipping interstitial generation with
interstitial_gen_kwargs=False, or using modified interstitial generation constraints (withinterstitial_gen_kwargs), or providing a list of known interstitial sites withinterstitial_coords.
Parsing
For defect calculation parsing, this can be slowed down in the case of large supercells, due to the large
sizes of the output vasprun.xml(.gz) files. Again, doped has been heavily-optimised to expedite this
process, and will use multiprocessing by default to accelerate when multiple CPUs are available. The main
bottleneck here is the loading and parsing of vasprun.xml(.gz) files. Parsing only has to be run once
however, and we encourage the saving of parsed outputs to json.gz files as shown in the tutorials
(and automatically performed by DefectsParser). Future improvements in the
efficiency of the pymatgen Vasprun parser for large files would be
very beneficial here.
Difficult Structural Relaxations & Energy/Force Convergence Issues
If defect supercell relaxations do not converge after multiple continuation calculations
(i.e. cp-ing CONTCAR to POSCAR and resubmitting the job), this is likely due to small
residual forces causing the local optimisation algorithm to struggle to find a solution, an error in the
underlying calculation and/or extreme forces.
If the calculation outputs show that the relaxation is proceeding fine, without any errors, just not converging to completion, then it suggests that the structure relaxation is bouncing around a narrow region of the potential energy surface. Here, the gradient-based geometry optimiser is struggling to converge.
Often (but not always) this indicates that the structure may be stuck around a saddle point or shallow local minimum on the potential energy surface (PES), so it’s important to make sure that you have performed structure-searching (PES scanning) with an approach such as ShakeNBreak (
SnB) to avoid this. You may want to try ‘rattling’ the structure to break symmetry in case this is an issue, as detailed in this part of theSnBdocs.Alternatively (if you have already performed SnB structure-searching), convergence of the forces can be aided by:
Switching the ionic relaxation algorithm back and forth (i.e. change
IBRIONto1or3and back).Reducing the ionic step width (e.g. change
POTIMto0.02in theINCAR)Switching the electronic minimisation algorithm (e.g. change
ALGOtoAllandISEARCHto1), if electronic convergence seems to be causing issues.Tightening/reducing the electronic convergence criterion (e.g. change
EDIFFto1e-7)Switching to reciprocal space projection operators (e.g. change
LREALtoFalse) can give slightly more accurate forces and avoid some convergence issues, but note that this can cause a systematic energy shift (typically on the order of 1 meV/atom), and so if switching this for relaxation convergence, a final total-energy calculation should be performed with the original settings (consistent with the bulk/other defect calculations) to obtain reliable relative (formation) energies.
If instead the calculation is crashing due to an error and/or extreme forces, a common culprit is the
EDWAVerror in the output file, which can often be avoided by reducingNCOREand/orKPAR. If this doesn’t fix it, switching the electronic minimisation algorithm (e.g. changeALGOtoAllandISEARCHto1) can sometimes help.If some relaxations are still not converging after multiple continuations, you should check the calculation output files to see if this requires fixing. Often this may require changing a specific
INCARsetting, and using the updated setting(s) for any other relaxations that are also struggling to converge.
ShakeNBreak
For tips on the ShakeNBreak part of the defect calculation workflow, please refer to the
ShakeNBreak documentation.
Competing Phases & Chemical Potentials
Accurate chemical potential limits require well-converged total energies for the host and all competing
phases (see the CompetingPhases / CompetingPhasesAnalyzer classes and the
chemical potentials tutorial). doped automates k-point
convergence testing and input file generation for these calculations, setting sensible defaults for
smearing/magnetisation (ISMEAR/SIGMA/NUPDOWN in VASP) etc. depending on whether a phase is
a metal, semiconductor/insulator or molecule. A few tips for keeping these calculations efficient –
particularly for metals, which are often the most expensive competing phases – are given below:
Pre-screen polymorphs without SOC: While spin-orbit coupling (SOC) can be important for accurate absolute energies in heavy-element systems (as discussed in Guidelines for robust and reproducible point defect simulations in crystals, Box 1), relative energies of polymorphs of the same composition/oxidation states` are usually reliable without SOC. Thus for systems with many candidate polymorphs, you can cheaply pre-screen with non-SOC calculations, and then only perform the (expensive) SOC calculations on the ground-state polymorph of each competing phase.
Smearing: Gaussian and Methfessel-Paxton smearing (
ISMEAR = 0and2) are thedopeddefault settings for insulators and metals for geometry relaxations, but tetrahedron smearing (ISMEAR = -5) typically gives more accurate total energies for the final single-point calculation and converges at lower k-point densities. This can be particularly useful for metallic competing phases, which require high k-point densities and thus can be very expensive, particularly if also using hybrid DFT and/or including spin-orbit coupling (SOC). It can be worth re-running k-point convergence testing (withwrite_kpoint_convergence_files()) usingISMEAR = -5to check for cheaper converged k-point densities for these final single-point calculations.``NKRED`` with hybrid DFT: The Fock exchange contribution in hybrid DFT typically converges at lower k-point densities than the other DFT energy terms, so the NKRED(X,Y,Z)
INCARinVASP(or nqx1/2/3 inQuantum ESPRESSO) can be used to reduce the number of k-points used for the Fock exchange to greatly reduce the cost with minimal loss of accuracy – as encouraged in the defect supercell workflow (see the VASP input file generation section of the defect generation tutorial, and thevasp_nkred_stdproperty). This is particularly useful for metallic competing phases, which require high k-point densities and thus can be very expensive/memory-demanding, particularly if also using hybrid DFT and/or including spin-orbit coupling (SOC), but can often use reduced Fock exchange k-point densities without significant loss of accuracy. Of course, if using results from calculations with reduced Fock exchange k-point densities, it’s important to check the accuracy of this approximation for your system (e.g. by running some quick accuracy/convergence tests).Note however that
NKRED(or alternativelyODDONLY/EVENONLY) must divide the number of k-points in each direction, so they can sometimes be awkward to use with symmetry-onvasp_stdcalculations (where the irreducible k-point grid isn’t set beforehand).
Memory vs k-points: Because QM/DFT calculations for metals typically need dense k-meshes, memory usage can sometimes become the limiting factor (particularly if using hybrid DFT and/or SOC). In some cases, it can be preferable to use a larger supercell of the phase, with a correspondingly reduced k-point grid (rather than a small cell with a very dense mesh) to help stay within memory limits.
Layered / Low Dimensional Materials
Layered and low-dimensional materials introduce complications for defect analysis. One point is that typically such lower-symmetry materials exhibit higher rates of energy-lowering defect reconstructions (e.g. 4-electron negative-U centres in Sb₂Se₃, vacancies in low-dimensional chalcogenides etc), as a result of having more complex energy landscapes, and so use of structure-searching strategies like ShakeNBreak can be particularly important.
Another is that often the application of charge correction schemes to supercell calculations with layered
materials may require some fine-tuning for converged results. To illustrate, for Sb₂Si₂Te₆ (
a promising layered thermoelectric material),
when parsing the intrinsic defects, the -3 charge antimony vacancy (v_Sb-3) gave this warning:
Estimated error in the Kumagai (eFNV) charge correction for defect v_Sb_-3 is 0.067 eV (i.e. which
is greater than the ``error_tolerance``: 0.050 eV). You may want to check the accuracy of the
correction by plotting the site potential differences (using
``defect_entry.get_kumagai_correction()`` with ``plot=True``). Large errors are often due to
unstable or shallow defect charge states (which can't be accurately modelled with the supercell
approach). If this error is not acceptable, you may need to use a larger supercell for more
accurate energies.
Note
Charge correction errors are estimated by computing the standard error of the mean of the electrostatic potential difference between the bulk and defect supercells, in the sampling region (far from the defect site), and multiplying by the defect charge. This gives a lower bound estimate of the true error in the charge correction for a given supercell.
Following the advice in the warning, we use the DefectEntry
get_kumagai_correction() method with plot=True to plot the site
potential differences for the defect supercell (which is used to obtain the eFNV (Kumagai-Oba) anisotropic
charge correction):
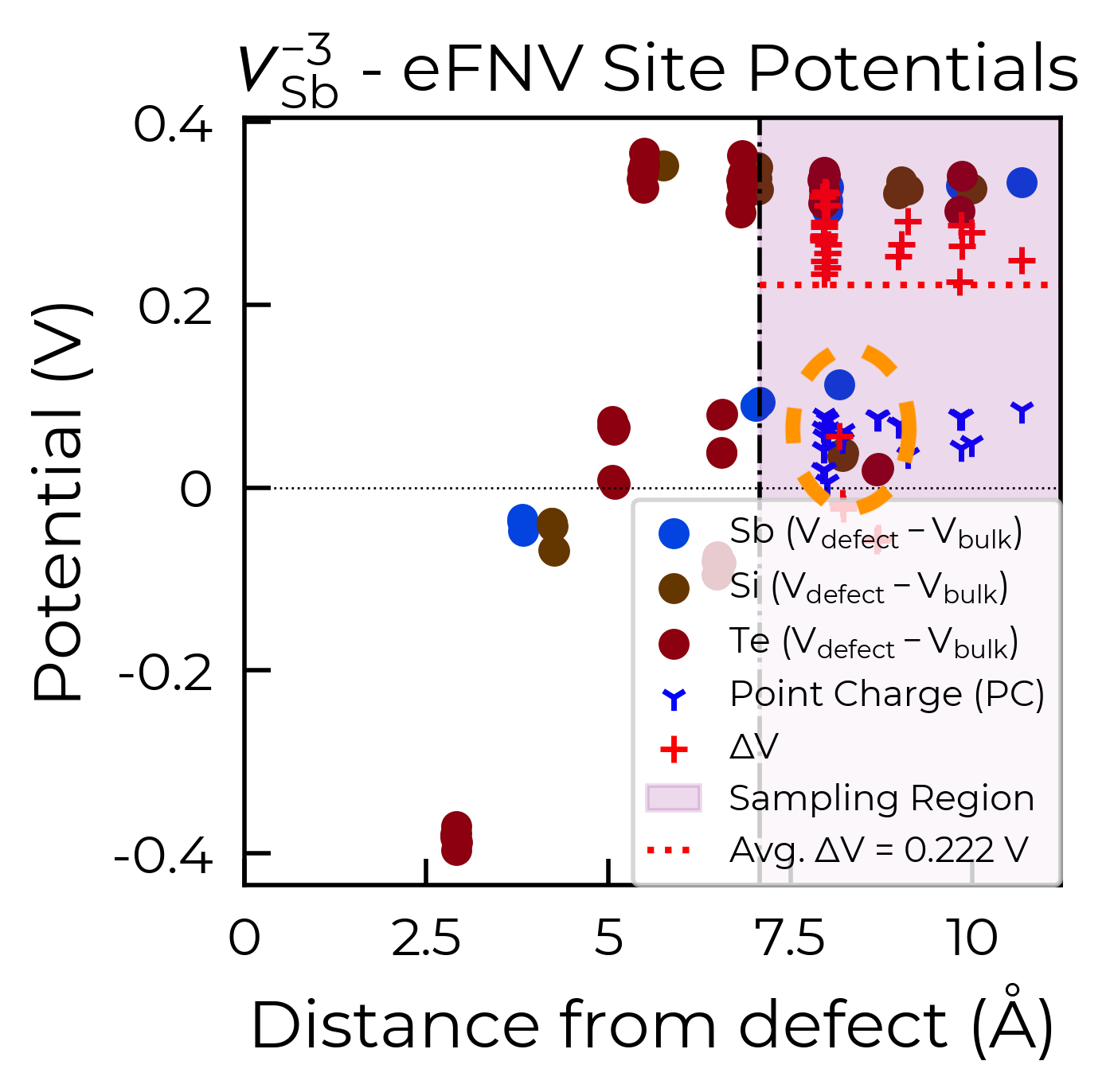
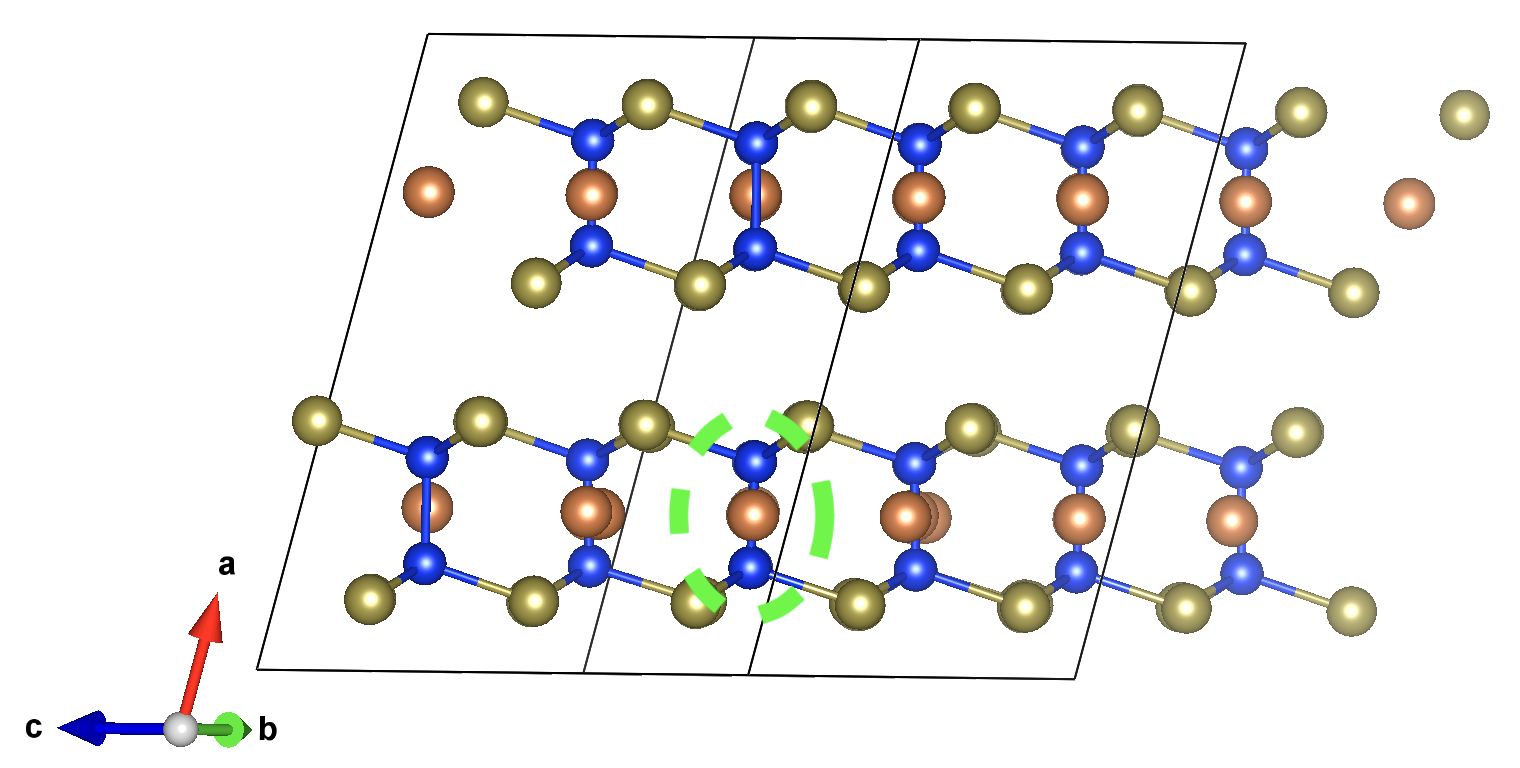
From the eFNV plot, we can see that there appears to be two distinct sets of site potentials, with one curving up from ~-0.4 V to ~0.1 V, and another mostly constant set at ~0.3 V. We can understand this by considering the structure of our defect (shown on the right), where the location of the Sb vacancy (hidden by the projection along the plane) is circled in green – we can see the displacement of the Sb atoms on either side.
Due to the layered structure, the charge and strain associated with the defect is mostly confined to the defective layer, while that of the layer away from the defect mostly experiences the typical long-range electostatic potential of the defect charge. The same behaviour can be seen for h-BN in the original eFNV paper (Figure 4d). This means that our usual default of using the Wigner-Seitz radius to determine the sampling region is not as good, as it’s including sites in the defective layer (circled in orange) which are causing the variance in the potential offset (ΔV) and thus the error in the charge correction.
To fix this, we can use the optional defect_region_radius or excluded_indices parameters in
DefectEntry get_kumagai_correction(), to exclude those
points from the sampling. For defect_region_radius, we can just set this to 8.75 Å here to avoid those
sites in the defective layer. Often it may not be so simple to exclude the intra-layer sites in this way
(depending on the supercell), and so alternatively we can use excluded_indices for more fine-grained
control. As we can see in the structure image above, the a lattice vector is aligned along the
inter-layer direction, so we can determine the intra-layer sites using the fractional coordinates of the
defect site along a:
# get indices of sites within 0.2 fractional coordinates along a of the defect site
sites_in_layer = [
i for i, site in enumerate(defect_entry.defect_supercell)
if abs(site.frac_coords[0] - defect_entry.defect_supercell_site.frac_coords[0]) < 0.2
]
correction, fig = dp.defect_dict["v_Sb-3"].get_kumagai_correction(
excluded_indices=sites_in_layer, plot=True
) # note that this updates the DefectEntry.corrections value, so the updated correction
# is used in later formation energy / concentration calculations
Below are the two resulting charge correction plots (using defect_region_radius on the left, and
excluded_indices on the right):
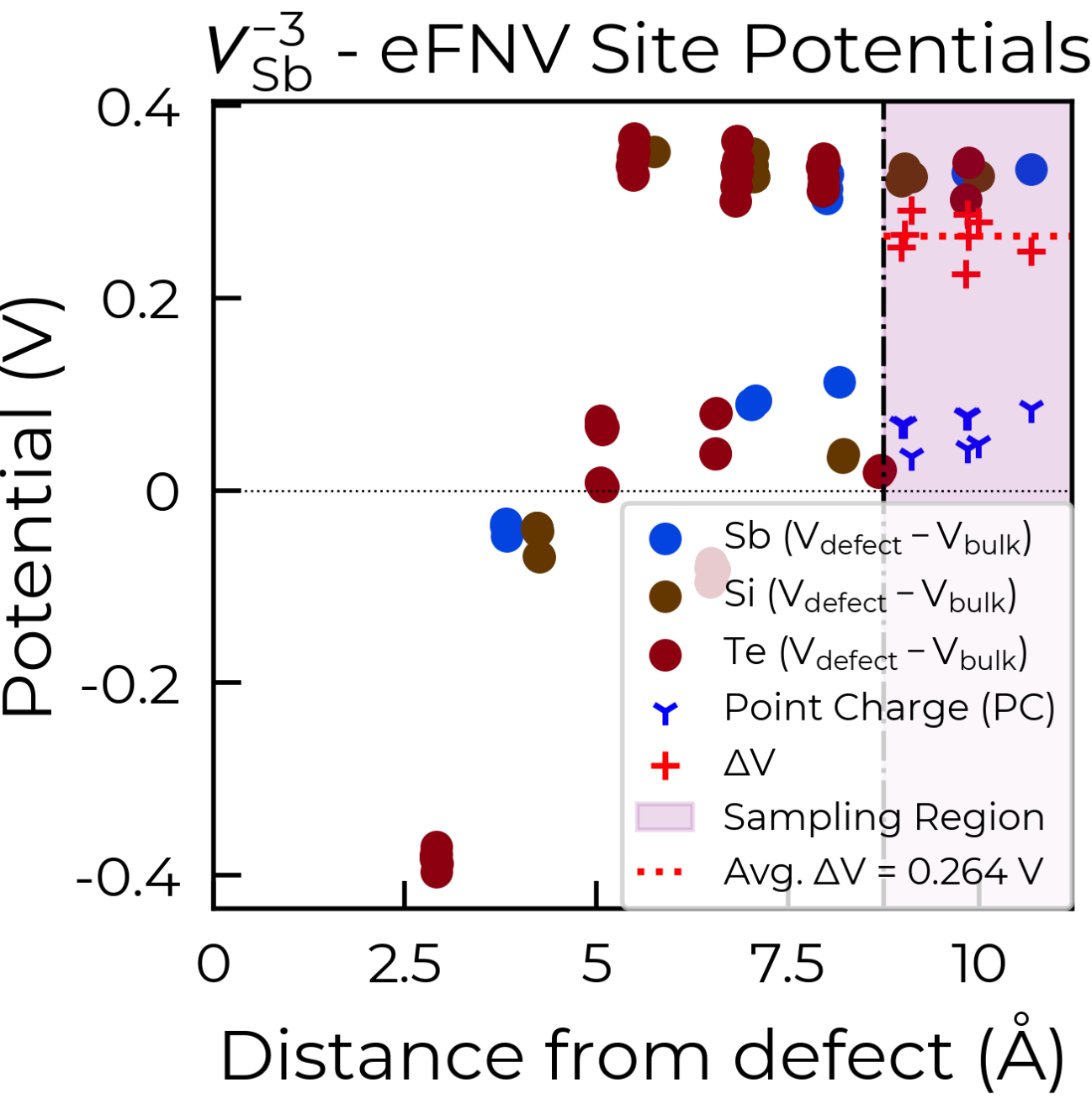
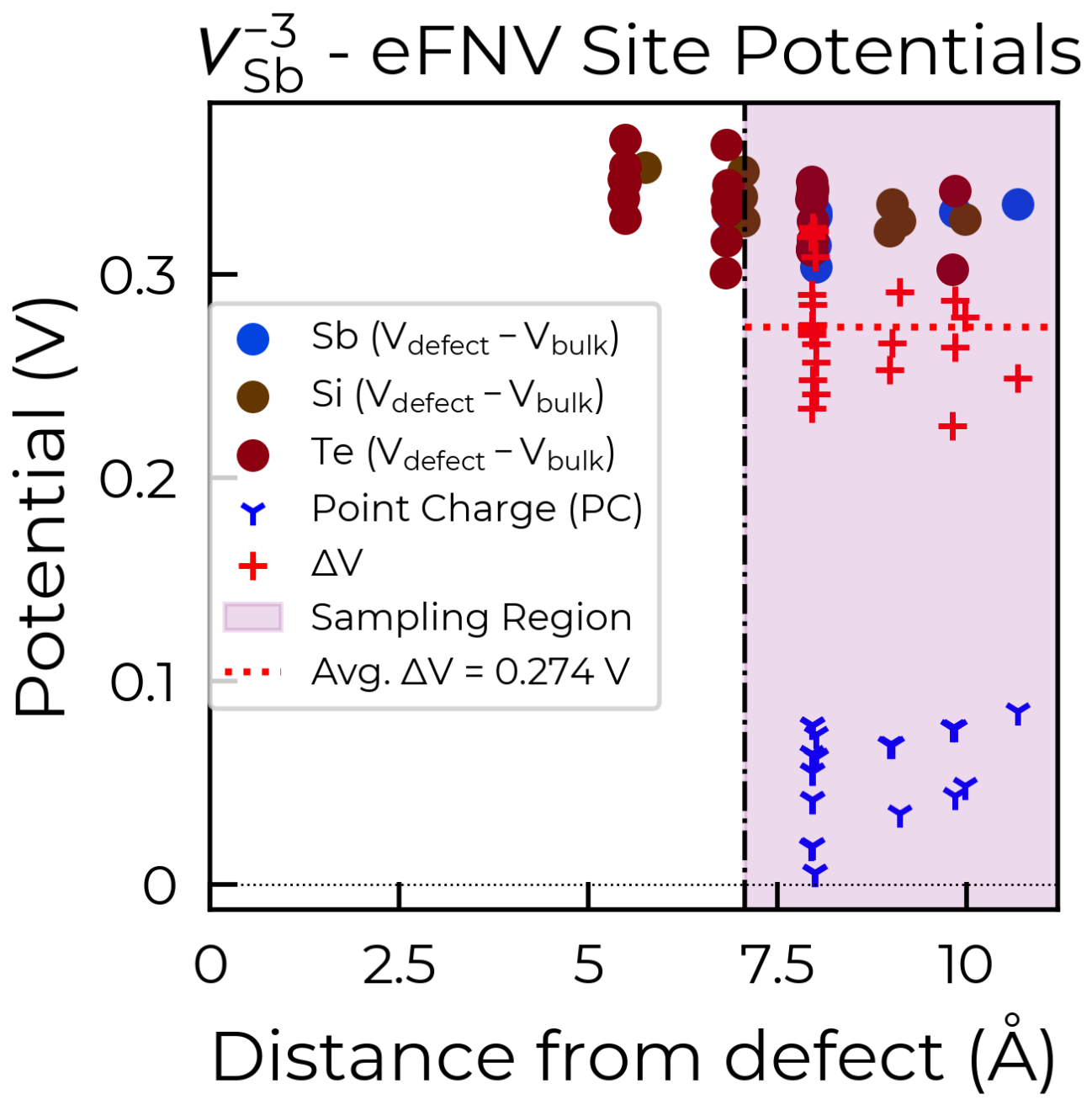
Tip
In other cases, if the error in the finite-size corrections are too large, we may need to compute the
given defect in a larger supercell. The stenciling functions in doped, which allow the
re-generation of (relaxed) defect structures in arbitrary supercells, can be very useful in these
cases. See the stenciling tutorial for examples and discussion.
Tip
Given that the magnitude of finite-size effects can vary dramatically by direction in layered/2D/highly-anisotropic materials (as discussed here), often the optimal supercell for these cases can differ from that returned by the default supercell generation algorithm (which optimises the minimum image distance for the minimum number of atoms), with e.g. a dielectric-weighted supercell generation approach often being more appropriate. See the dielectric-weighted supercell generation section of the advanced analysis tutorial for an example and discussion.
2D Materials and Surface Defects
Following on from the layered materials discussion above, there are additional complications when modelling defects in two-dimensional materials, or defects near surfaces in three-dimensional materials, primarily related to the implementation of finite-size charge corrections. For a detailed discussion, see Kumagai Phys Rev B 2024.
In these cases, defects can be generated using a similar workflow as for 3D materials, where now our input
host system to DefectsGenerator should be a slab structure with a converged
vacuum size. doped will automatically generate all symmetry-inequivalent defects in this slab, and
relevant properties such as distance to surface can be readily calculated through the site information and
pymatgen Structures stored with the doped DefectEntry / Defect objects.
The calculation inputs can then be generated as before, along with ShakeNBreak distortions
(recommended), and then parsed with doped’s DefectsParser class similar to 3D
materials.
For charge corrections however, it is recommended to use the advanced finite-size corrections adapted for 2D materials (to correctly model the distance-dependent dielectric profile in vacuum). Possible approaches include that of Noh et al. and Komsa et al. (“NK” method), now implemented in pydefect_2d; the approach of Freysoldt and Neugebauer, implemented in sxdefectalign2d; or the self-consistent potential correction (SCPC) method introduced by da Silva et al. with implementation details here.
Thus it is recommended to set skip_corrections = False when parsing defects in 2D materials / surface
defects with DefectsParser (to avoid automatic application of 3D charge
corrections), and then update the DefectEntry charge corrections with the
externally-calculated 2D values, before continuing your analyses. For example, the
DefectEntry charge corrections can be updated like this:
# updated charge corrections using an appropriate 2D materials scheme
new_corrections = { # format: {entry_name: correction in eV}
"v_Cd_-1": +0.2,
"v_Cd_-2": +0.4,
"Te_Cd_+1": -0.2,
} # note that these corrections are added to the un-corrected formation energies, to give the final values
for entry_name, entry in defect_thermo.defect_entries.items():
if entry_name in new_corrections:
for corr_key in list(entry.corrections.keys()): # this inner loop is not required if ``skip_corrections`` was set to ``True``
if "charge_correction" in corr_key:
print(f"Removing {corr_key} for {entry_name}: {entry.corrections.pop(corr_key):+.2f} eV")
entry.corrections.update({"2D Correction": new_corrections[entry_name]})
print(f"New correction for {entry_name}: {new_corrections[entry_name]:+.2f} eV")
else:
print(f"No corrections to add for {entry_name}")
Eigenvalue / Electronic Structure Analysis
In doped, we can use the DefectEntry
get_eigenvalue_analysis() method to analyse the orbital character and
localisation of single-particle eigenstates from the underlying electronic structure calculations. For this,
we employ the methodology of
Kumagai et al. (through an interface with
pydefect), which allows in-depth analysis of localised/deep in-gap defect states and their effects on
the band edges, as well as the automated identification of shallow / perturbed host states (PHS) – see
the following section for an example of this analysis. The
easyunfold package for band structure unfolding can also be
quite useful for extending this electronic structure analysis.
The optional argument parse_projected_eigen in DefectsParser (True by
default) controls whether to load the projected eigenvalues & orbitals, which then allows
get_eigenvalue_analysis() to be called – returning information about the
nature of the band edge and in-gap states, allowing defect states (and whether they are deep or
shallow/PHS) to be automatically identified and characterised.
Furthermore, a plot of the single-particle electronic eigenvalues is returned (if plot = True;
default). Note that for VASP to output the necessary data for this analysis, your INCAR file needs to
include LORBIT > 10 (to obtain the projected orbitals).
In the examples below (both of which are shown in the advanced analysis tutorial), we plot the single particle levels for the cadmium vacancy in CdTe (VCd) in each of its charge states (0, -1 and -2); calculated with spin-orbit coupling (SOC) and a 2x2x2 k-point mesh:
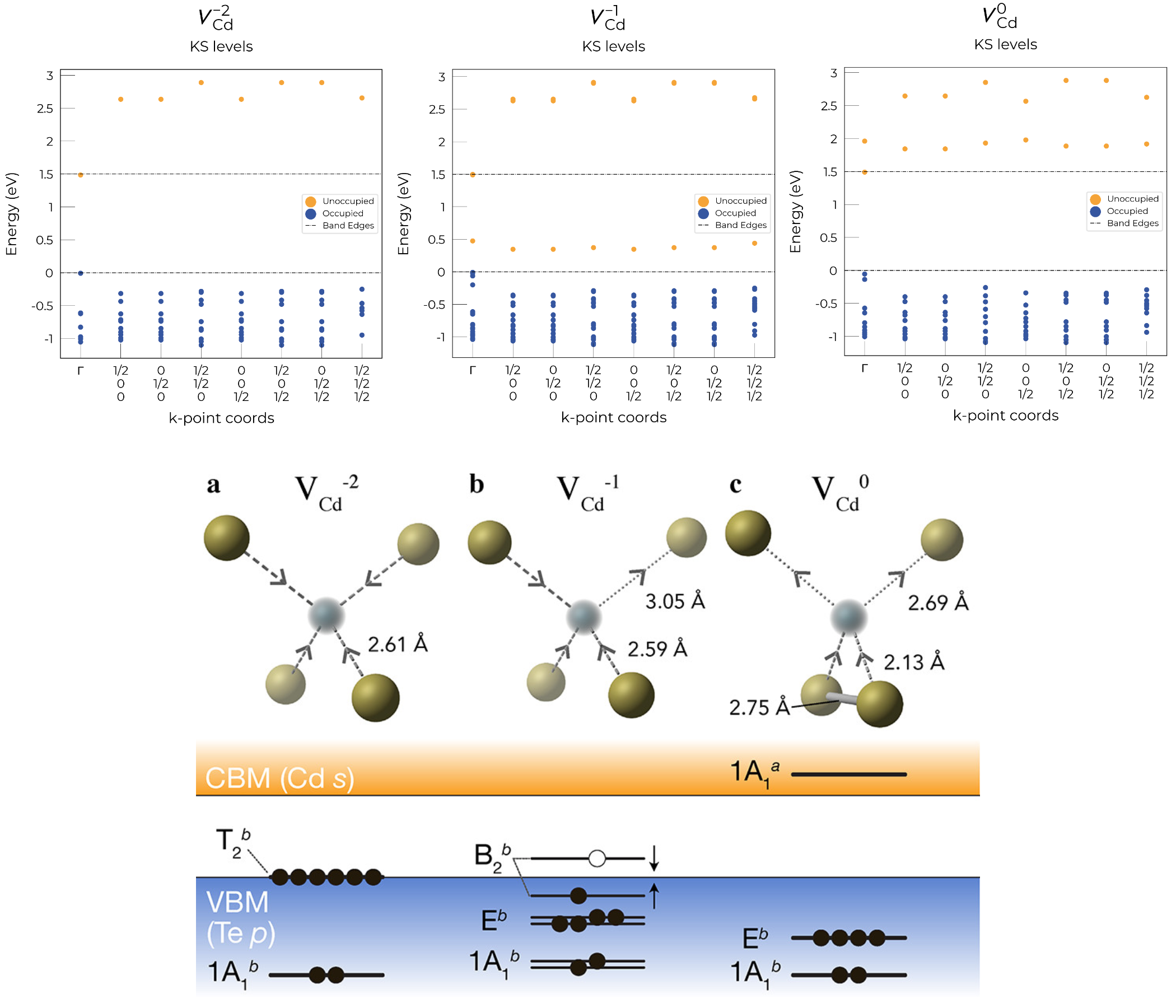
Here we can see that these plots nicely match the schematic depiction from this paper on vacancies in CdTe, where we have no in-gap states for the fully-ionised VCd-2 as expected, an in-gap hole polaron state for VCd-1, and an anti-bonding dimer state for VCd0 just above the CBM.
Perturbed Host States (Shallow Defects)
One of the most common reasons for performing this electronic structure analysis is to identify and analyse shallow defect states. Certain point defects form shallow (hydrogen-like) donor or acceptor states, known as perturbed host states (PHS). These states typically have wavefunctions distributed over many unit cells in real space, requiring exceptionally large supercells or dense reciprocal space sampling to properly capture their physics (see this review or this paper). This weak attraction of the electron/hole to the defect site corresponds to a relatively small donor/acceptor binding energy (i.e. energetic separation of the corresponding charge transition level to the nearby band edge), which is typically <100 meV.
As discussed in this Guidelines for Defect Simulations
perspective, current supercell correction schemes cannot accurately account for finite-size errors obtained
when calculating the energies of PHS (shallow defect states) in moderate supercells, so it is recommended
to denote such shallow defects as PHS and conclude only qualitatively that their transition level is
located near the corresponding band edge. An example of this is given in
Kikuchi et al. Chem. Mater. 2020.
As discussed below, this is performed automatically in doped.
Tip
Typically, the shallow defect binding energy can be reasonably well estimated using the hydrogenic model, similar to the Wannier-Mott exciton model, which predicts a binding energy given by:
where \(\bar{m}\) is the harmonic mean (i.e. conductivity) effective mass of the relevant
charge-carrier (electron/hole), \(\epsilon\) is the total dielectric constant
(\(\epsilon = \epsilon_{\text{ionic}} + \epsilon_{\infty}\)) and Ry is the Rydberg constant
(i.e. binding energy of an electron in a hydrogen atom; ~13.6 eV).
This formula is used in the shallow_dopant_binding_energy() convenience
function, with example usage shown
here in the advanced analysis tutorial.
As shown in the tutorial example, this formula can also be used to estimate the Wannier-Mott exciton
binding energy, when the reduced mass of the electron-hole pair is used for the effective mass.
As discussed in the section above, we employ the methodology of Kumagai et al. to analyse the orbital character and localisation of single-particle eigenstates from the underlying electronic structure calculations, which allows the automated identification of shallow states.
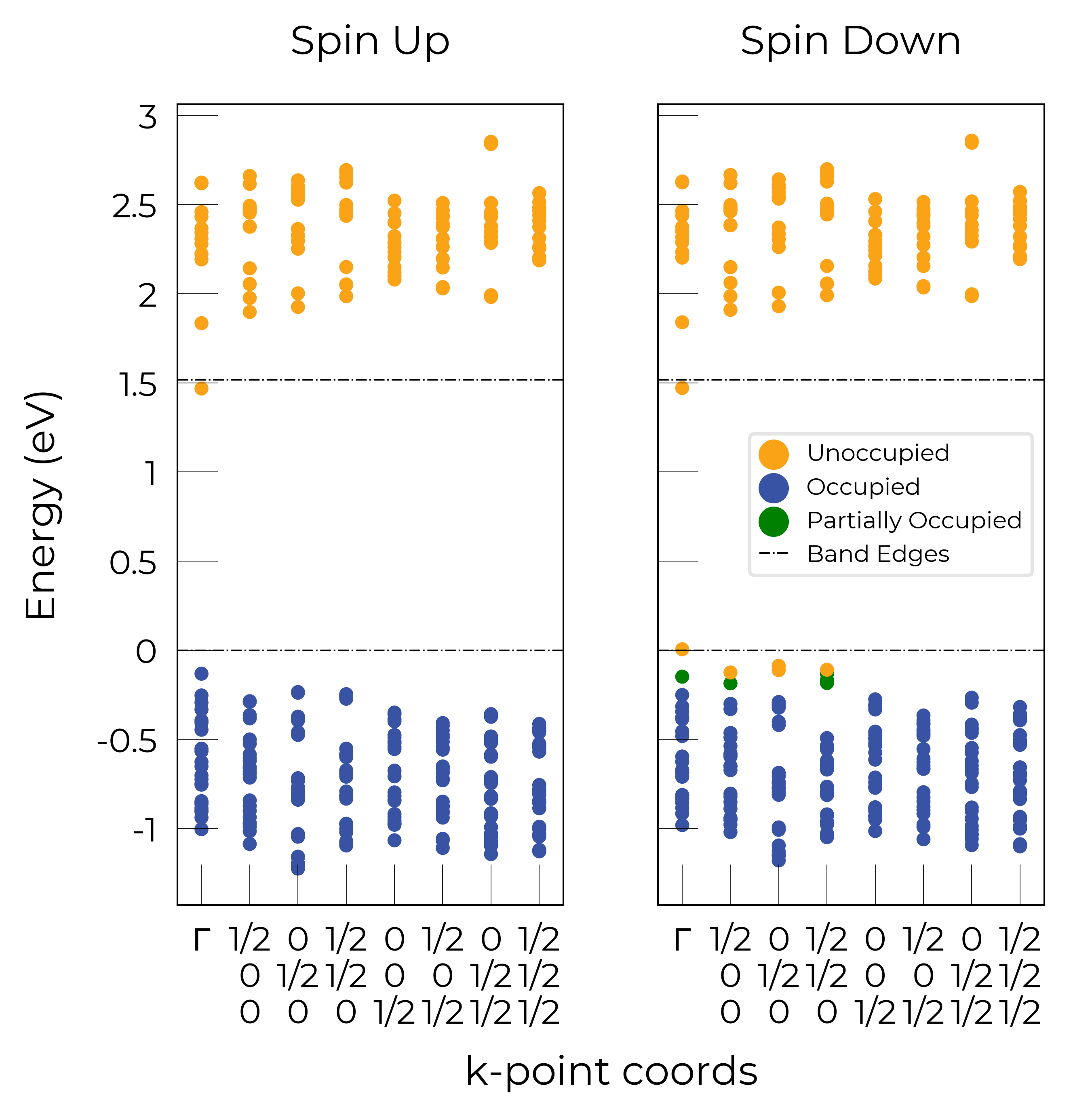
In the example below, the neutral copper vacancy in Cu₂SiSe₃ was determined to be a PHS. This was additionally confirmed by performing calculations in larger supercells and plotting the charge density. Important terms include:
P-ratio: The ratio of the summed projected orbital contributions of the defect & neighbouring sites to the total sum of orbital contributions from all atoms to that electronic state. A value close to 1 indicates a localised state.Occupation: Occupation of the electronic state / orbital.vbm has acceptor phs/cbm has donor phs: Whether a PHS has been automatically identified. Depends on how VBM-like/CBM-like the defect states are and the occupancy of the state.(X vs. 0.2)refers to the hole/electron occupancy at the band edge vs the default threshold of 0.2 for flagging as a PHS (but you should use your own judgement of course).Localized Orbital(s): Information about localised defect states, if present.
Additionally, Index refers to the band/eigenvalue index in the QM/DFT calculation, Energy is its
eigenvalue energy at the given K-point coords, Orbitals lists the projected orbital contributions
to that state, and OrbDiff is the normalised difference in projected orbital contributions to the
VBM/CBM states between the bulk and defect supercells.
bulk = "Cu2SiSe3/bulk/vasp_std"
defect = "Cu2SiSe3/v_Cu_0/vasp_std/"
dielectric = [[8.73, 0, -0.48],[0., 7.78, 0],[-0.48, 0, 10.11]]
defect_entry = DefectParser.from_paths(defect, bulk, dielectric).defect_entry
bes, fig = defect_entry.get_eigenvalue_analysis()
print(bes) # print information about the defect state
-- band-edge states info
Spin-up
Index Energy P-ratio Occupation OrbDiff Orbitals K-point coords
VBM 347 3.539 0.05 1.00 0.01 Cu-d: 0.35, Se-p: 0.36 ( 0.000, 0.000, 0.000)
CBM 348 5.139 0.03 0.00 0.03 Se-s: 0.20, Se-p: 0.11, Si-s: 0.13 ( 0.000, 0.000, 0.000)
vbm has acceptor phs: False (0.000 vs. 0.2)
cbm has donor phs: False (0.000 vs. 0.2)
---
Localized Orbital(s)
Index Energy P-ratio Occupation Orbitals
Spin-down
Index Energy P-ratio Occupation OrbDiff Orbitals K-point coords
VBM 347 3.677 0.06 0.00 0.01 Cu-d: 0.34, Se-p: 0.35 ( 0.000, 0.000, 0.000)
CBM 348 5.142 0.04 0.00 0.03 Se-s: 0.20, Se-p: 0.11, Si-s: 0.13 ( 0.000, 0.000, 0.000)
vbm has acceptor phs: True (1.000 vs. 0.2)
cbm has donor phs: False (0.000 vs. 0.2)
---
Localized Orbital(s)
Index Energy P-ratio Occupation Orbitals
The plot of the single particle levels is shown below (left), and an example of how you might chose to represent the PHS on the transition level diagram with a clear circle is shown on the right.
Note
The classification of electronic states as band edges or localized orbitals is based on the similarity
of orbital projections and eigenvalues between the defect and bulk cell calculations (see
the DefectEntry get_eigenvalue_analysis()
docstring). You may want to adjust the default values of the similar_orb/energy_criterion keyword
arguments, as the defaults may not be appropriate in all cases. In particular, the P-ratio values can
give useful insight, revealing the level of (de)localisation of the states. It is recommended to
additionally manually check the real-space charge density (i.e. PARCHG) of the defect state when
possible, to confirm the identification of a PHS.
Note
As mentioned above, the eigenvalue analysis functions use code from pydefect, so please cite the
pydefect paper if using these analyses in your work:
“Insights into oxygen vacancies from high-throughput first-principles calculations” Yu Kumagai, Naoki Tsunoda, Akira Takahashi, and Fumiyasu Oba Phys. Rev. Materials 5, 123803 (2021) – 10.1103/PhysRevMaterials.5.123803
In doped, this eigenvalue analysis is performed automatically, and shallow/unstable defect charge
states can be omitted from plotting and analysis using the unstable_entries argument and/or
the DefectThermodynamics
prune_to_stable_entries() method. By default, parsed
defect entries which are detected to be shallow (‘perturbed host’) states and unstable for Fermi levels in
the band gap are omitted from plotting for clarity & accuracy.
Density of States (DOS) Calculations
As discussed in the
thermodynamics & doping tutorial,
the electronic density of states (DOS) of the bulk material (provided as bulk_dos) is required to
determine carrier concentrations and thus (quasi-)equilibrium Fermi levels and defect concentrations under
various conditions, along with the defect formation energies and other inputs.
The bulk DOS (bulk_dos) calculation should be a static calculation with the primitive unit cell, using:
Dense k-point sampling and energy grid spacing (
KPOINTSandNEDOSrespectively withVASP) to give a converged DOS spectrum (see e.g. vaspup2.0) and an accurate band gap.Ideally tetrahedron smearing (
ISMEAR = -5inVASP) for improved DOS convergence with respect to k-points.Consistent QM/DFT functional settings as used for the final bulk/defect supercell calculations (e.g.
LHFCALC,AEXX,LSORBITetc).The same host crystal structure used to generate the bulk/defect supercells, and same pseudopotentials (
POTCARs inVASP) used for the supercell calculations.
See the MgO GGA workflow tutorial for an example of setting up a bulk DOS calculation.
If there is a significant mismatch between the VBM eigenvalue or band gap of the bulk DOS (bulk_dos)
and bulk/defect supercell calculations (stored as the
vbm/
gap attributes of
DefectThermodynamics), this can lead to inaccuracies in the
thermodynamics & concentration analyses. If doped detects this to be the case, it will throw a warning
like: The band gap / VBM eigenvalue of the bulk DOS calculation (... eV) differs by >0.05 eV from
DefectThermodynamics.vbm/gap....
This can arise for a number of reasons:
Differences in DFT functional choices, pseudopotentials (
POTCARs inVASP) or bulk crystal structure/volume.This can cause severe errors and should be rectified so that the DOS and supercell calculation settings are made consistent as discussed above.
Effects of smearing schemes (e.g.
ISMEARinVASP), such that the band edges are not accurately determined in the bulk DOS calculation.If possible, use tetrahedron smearing (
ISMEAR = -5) for improved DOS convergence with respect to k-points as discussed above. If this is not possible, but dense k-point sampling and energy grid spacing is being used, then this should not significantly impact accuracies and the warning can be ignored (setskip_dos_check = Trueto silence the warning).
Significant differences in k-point sampling between the bulk DOS and supercell calculations, such that the same band edges are not captured in both calculations. This can happen in cases where band edges occur at k-points which are not high-symmetry and/or are outside of the k-point grid of the supercell calculations.
Assuming the k-point sampling of the DOS calculation is sufficiently dense, then the issue is just that the VBM eigenvalue and band gap of the bulk supercell calculation is not accurate. This can be rectified by using the
bulk_band_gap_vroption during defect parsing (seeDefectsParserdocstring) to set the bulk band gap and VBM eigenvalue to the correct values.In this case, the absolute values of predictions should not be affected as the eigenvalue references in the calculations are consistent, just the reported Fermi levels will be referenced to the
vbmattribute which may not be the exact VBM position here.
Small numerical inaccuracies in determining the VBM eigenvalue or band gap directly from the DOS spectrum.
Determining the VBM eigenvalue or band gap directly from a DOS spectrum is not entirely straightforward, due to effects of smearing, noise, finite sampling etc. If all other possible issues above have been ruled out and the detected mismatch is relatively small, then this may be the case and can be ignored (set
skip_dos_check = Trueto silence the warning) — accuracies should not be significantly affected.
Note
The Fermi level will be always referenced to the vbm
attribute of DefectThermodynamics.
Magnetization
Proper accounting of magnetization and spin multiplicity is important for accurate defect calculations and
analysis. For defect species with odd numbers of electrons (and thus being open-shell), they will adopt
non-zero integer spin states, while defect species with even numbers of electrons can be either
closed-shell (spin-paired) or open-shell (spin-active), depending on the defect species and its electronic
structure. As such, defect calculations should typically be performed with spin polarisation allowed in all
cases (i.e. with ISPIN = 2 or LSORBIT = True (for SOC) in VASP).
Tip
If we have (nearly) converged the geometry relaxation for an even-electron defect species and there is
no non-zero magnetic moments on any site (given by the magnetization output in the OUTCAR file)
– and so adopting a closed-shell electronic structure, then we can set ISPIN = 1 (turning off
spin polarisation) for subsequent calculations to reduce the computational cost.
The snb-mag --verbose CLI command from ShakeNBreak can be used to automatically check the
magnetization of a VASP defect calculation in this way (and is automatically used by snb-run to
set ISPIN = 1 for continued ShakeNBreak relaxations of any closed-shell defect calculations,
if it is being used to manage the structure-searching calculations).
❯ snb-mag -h
Usage: snb-mag [OPTIONS]
Checks if the magnetization (spin polarisation) values of all atoms in the
VASP calculation are below a certain threshold, by pulling this data from
the OUTCAR. Returns a shell exit status of 0 if magnetization is below the
threshold and 1 if above.
Options:
-o, --outcar FILE Path to OUTCAR(.gz) file
-t, --threshold FLOAT Atoms with absolute magnetization below this value
are considered un-magnetised / non-spin-polarised.
The threshold for total magnetization is 10x this
value. [default: 0.01]
-v, --verbose Print information about the magnetization of the
system.
-h, --help Show this message and exit.
In most cases and particularly for s/p orbital systems, odd electron defects will adopt a doublet spin
state (S = 1/2, one unpaired electron), while even electron defects will tend to adopt a closed-shell
singlet spin state (S = 0, no unpaired electrons), as a consequence of the Aufbau principle and Hund’s
rule. For input file generation in VASP, NUPDOWN is used in the INCAR to enforce the total
magnetization (number of unpaired electron spins), which by default is set to 0 for even-electron
and 1 for odd-electron defect species in doped.
However, this is not always the case and often we can have open-shell triplet states for
even-electron defects (with S = 1, two unpaired electrons) or quartet states for odd-electron defects
(with S = 3/2, three unpaired electrons). Such cases are most common when the defect species adopts a
bipolaron/multi-polaron state (e.g. for VCd0* in
CdTe, VSe0 in
t-Se, or VP-1
in NaP), a molecular
dimer-like state (such as O2 species in
oxides or
carbon pairs in silicon) – for which doped
has an automated warning upon parsing, defects involving multiple localised d/f electrons, or
orbital-degenerate/correlated defects where Hund’s rule implies open-shell solutions (such as the
highly-studied NV centre in diamond
or transition metal impurities in silicon).
If you encounter defect states like these and/or suspect that alternative spin configurations may be
possible, you should test the different possibilities by setting NUPDOWN (and possibly MAGMOM,
discussed below) accordingly – ideally performing the full structure-searching calculations for these
species with these settings, as the potential energy surface can differ significantly under different spin
states.
Note
In general, it is best to explicitly specify the system spin state (i.e. with NUPDOWN) in QM/DFT
calculations, rather than leaving this as a free parameter, as not enforcing this constraint can often
lead to erroneous and unphysical results in the form of partial orbital occupation and spins. This
can occur because the DFT self-interaction error initially favours delocalisation of the unpaired
electron density, and converges to this unphysical result.
As well as setting the total spin state of our supercell with NUPDOWN, another parameter that can be
important in certain cases is the individual site magnetic moments, which can be initialised with the
MAGMOM tag in the INCAR (see the VASPwiki page).
This tag is not set by default in doped, using the VASP default initialisation of
MAGMOM = NIONS*1.
This tag is particularly important for magnetic materials (as discussed in the
Magnetism section of the
ShakeNBreak tips page), and can be useful if trying to favour a specific polaron/spin configuration
(as briefly discussed at this point in the
YouTube defects tutorial). This tag can be set using the user_incar_settings parameter in the
doped.vasp classes, for which the python API helps streamline this process when setting MAGMOM
for multiple defects.
Note
For magnetic competing phases, the spin configuration should also be appropriately set. doped will
automatically set ISPIN=2 (allowing spin polarisation) and NUPDOWN according to the
magnetization output from the Materials Project calculation of the competing phase, but MAGMOM
(and possibly ISPIN/NUPDOWN) may also need to be set to induce a specific spin configuration in
certain cases.
When parsing defect calculations, doped will automatically extract the total magnetization from the
VASP output files, in order to determine the spin multiplicity. This value is stored in
the degeneracy_factors DefectEntry attribute
(DefectEntry.degeneracy_factors["spin degeneracy"], and printed in the
DefectThermodynamics
get_symmetries_and_degeneracies() method) and used in
thermodynamic analyses. See the spin_degeneracy_from_vasprun() function for
details.
Symmetry Precision (symprec)
When computing the symmetries of structures, a threshold parameter has to be set in order to distinguish
structural/positional noise from distinct site differences. In doped as in spglib (and
pymatgen), this can be controlled with the symprec parameter (which can be set in
DefectsParser, DefectParser, all
DefectThermodynamics symmetry/concentration functions,
get_orientational_degeneracy(),
point_symmetry_from_defect_entry() and others).
By default, doped uses a value of symprec = 0.01 for unrelaxed/bulk structures (matching the
pymatgen default), and a larger symprec = 0.1 for determining the point symmetries (and thus
orientational degeneracies) of relaxed defect structures to account for residual structural noise.
This symprec value of 0.1 also matches that used by the Materials Project.
You may want to adjust symprec for your system (e.g. if there are very slight octahedral distortions
etc.).
Tip
Note that you can directly use the point_symmetry_from_structure()
function from doped.utils.symmetry to obtain the relaxed or unrelaxed (bulk site) point symmetries
of a given defect supercell, directly from just the relaxed structures, regardless of whether these
defects were generated/parsed with doped.
Note
Wyckoff letters for lattice sites can depend on the ordering of elements in the conventional standard
structure, for which doped uses the spglib convention (e.g. in the
DefectsGenerator info output).
Note
By default, magnetic symmetry (i.e. MAGMOMs) are not used in symmetry analysis in doped, as noise
in these values (particularly in structures from the Materials Project) often leads to incorrect
symmetry determinations. To use magnetic moments in symmetry analyses, set the environment variable
USE_MAGNETIC_SYMMETRY=1 (i.e. os.environ["USE_MAGNETIC_SYMMETRY"] = "1" in Python).
Open Science and Reproducibility
Robust, open and reproducible science greatly strengthens the impact of research. This is especially true for computational defect modelling, given the many steps and complexities involved – see Guidelines for robust and reproducible point defect simulations in crystals for discussion.
doped has been built to aid robustness and reproducibility for computational defect studies.
We highly recommend that the doped/ShakeNBreak class objects, which store key metadata
and can be directly output to lightweight json(.gz) files (such as
DefectThermodynamics, DefectsGenerator,
Distortions, CompetingPhasesAnalyzer) be
shared in open-access repositories (e.g. Zenodo, Materials Cloud, Figshare) upon publication, along with
relevant raw computational data. It is also helpful to use the summary functions such as the
get_formation_energies() and
get_symmetries_and_degeneracies() methods for
DefectThermodynamics,
the get_formation_energy_df(),
calculate_chempots() and
to_LaTeX_table() methods for
CompetingPhasesAnalyzer etc – which output pandas
DataFrames which can be output to csv (with .to_csv(), see tutorials) and imported to
Microsoft Word / converted to LaTeX (Tables Generator) – to
summarise key quantities in Supplementary Information
files.
Examples of these practices are shown in
Intrinsic point defect tolerance in selenium for indoor and tandem photovoltaics
& Defect Tolerance via External Passivation in the Photocatalyst SrTiO₃:Al.
Further details on serialisation and data provenance utilities in doped are given below.
Serialisation & Data Provenance (JSON/csv)
To aid calculation reproducibility, data provenance and easy sharing/comparison of pre- and post-processing
stages of the defect workflow, doped objects have been made fully serializable, meaning they can be
easily saved and (re-)loaded from compact, lightweight .json files. As demonstrated at
various stages in the tutorials, this can be achieved using the dumpfn/loadfn functions from
monty.serialization, or with the to_json()/
from_json() methods provided for the Defect, DefectEntry, DefectsGenerator
and DefectThermodynamics classes:
# save a DefectThermodynamics object to a JSON file
defect_thermo.to_json("MgO_DefectThermodynamics.json.gz")
# then later in a different python session or notebook, we can reload the
# DefectThermodynamics object from the JSON file, containing all the associated info
from doped.thermodynamics import DefectThermodynamics
defect_thermodynamics = DefectThermodynamics.from_json("MgO_DefectThermodynamics.json.gz")
# alternatively, we can directly use the monty dumpfn/loadfn functions
# directly on any doped object, e.g. with our ``DefectsSet`` object
# containing all the info on the generated VASP input files:
from monty.serialization import dumpfn, loadfn
dumpfn(obj=defects_set, fn="MgO_DefectsSet.json.gz")
# and again later reload the object from the JSON file
defects_set = loadfn("MgO_DefectsSet.json.gz")
Note
While these JSON files tend to have relatively small file sizes anyway, we can further reduce their
size by saving to / loading from gzip or bz2 compressed JSON files, by specifying
.json.gz/.json.z/.json.bz2 as the file extension in the serialization functions.
In the typical defect calculation workflow with doped (exemplified in the tutorials), the following
JSON files are automatically written to file:
The
DefectsGeneratorobject ordefect_entriesdictionary that is input toDefectsSet, when writingVASPinput files withDefectsSetwrite_files()– written tooutput_path. Additionally, for each calculation directory generated, the correspondingDefectEntryobject is written to a{DefectEntry.name}.jsonfile in the directory so that all information on the generated defect structure, charge state etc. is preserved in the calculation directory.The parsed defect entries dict (
defect_dict) when defect calculations are parsed withDefectsParser– written tooutput_path. The JSON filename can be set with e.g.DefectsParser(json_filename="custom_name.json"), but the default is{Host Chemical Formula}_defect_dict.json.gz.Additionally, if following the recommended structure-searching approach with
ShakeNBreakas shown in the tutorials,distortion_metadata.jsonfiles will be written to the top directory (output_path, containing distortion information about all defects) and to each defect directory (containing just the distortion information for that defect) when runningDistortions.write_vasp_files(output_path=".")(see theDistortionsclass docstring).
In most cases it is also recommended to save the DefectThermodynamics
object to file once generated (using the to_json()
method), to avoid having to re-parse at any later stage, however this is not done automatically.
DataFrame Outputs
Many analysis methods in doped return pandas DataFrame objects as the result, such as the
get_symmetries_and_degeneracies(),
get_formation_energies(),
get_equilibrium_concentrations(),
get_fermi_level_and_concentrations(),
get_dopability_limits(),
get_doping_windows() and
get_transition_levels() methods for DefectThermodynamics objects, and the
formation_energy_df attribute and
calculate_chempots() method for
CompetingPhasesAnalyzer. As mentioned in the tutorials, these
DataFrame objects can be output to csv (or json, xlsx etc., see the pandas API docs
here) using the to_csv/
to_json methods:
# save the formation energies DataFrame to a csv file
defect_thermo.get_formation_energies().to_csv("MgO_formation_energies.csv")
These csv files can easily be used as data tables when writing up results, by directly importing to
Microsoft Word or converting to LaTeX format using Tables Generator.
CompetingPhasesAnalyzer can also be reinitialised from a saved csv
formation energies file with the from_csv()
method.
Note
Have any tips for users from using doped? Please share it with the developers and we’ll add them here!